


Spike protein (inc vax) induced immunodeficiency & carcinogenesis megathread #25: T- helper 17 cells & interleukin 17

Any extracts used in the following article are for non commercial research and educational purposes only and may be subject to copyright from their respective owners.
Hat-tip to John Paul:
Reverse AIDS - Part IV
The roads to Rome
“You don’t need a severe case of Covid-19 for the stimulation of the necessary cytokines and everything else to produce enough quantities of specific cells and it’s proteins to keep shifting and causing transient immune dysfunction. The vaccinated encounter themselves in a perpetual state of this.”
https://worldedge.substack.com/p/reverse-aids-part-iv?s=r
Summary:
Interleukin: any of a class of glycoproteins produced by leucocytes for regulating immune responses.
Spike protein, whether viral or synthetic in origin due to transfection, promotes the secretion of inflammatory cytokines including interleukin 6 (IL-6) & tumor necrosis factor alpha (TNF-a) that can disrupt regulatory T cell differentiation by interfering with tumor growth factor beta (TGF-B), which then promotes T helper 17 cells (Th17) that secrete interleukin 17 (IL-17A).
In a normal response to invading pathogens or fungi this is healthy if limited, but if over promoted can either set off a feedback cascade of cytokines (cytokine storm) or lead to a dysfunctional immune response that can in the right context promote tumor growth or lead to autoimmune disorders, and suppress responses to other pathogens. Balance is the key here to avoiding histopathology.
IL-17A can be thought of as a key inflammatory cytokine at the heart of many pathophysiological pathways.
Background:
Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders (2010)
“Regulatory T cells (Tregs) are a specialized subpopulation of T cells that act to suppress immune response, thereby maintaining homeostasis and self-tolerance. It has been shown that Tregs are able to inhibit T cell proliferation and cytokine production and play a critical role in preventing autoimmunity.”
Th17 cells and Tregs: unlikely allies (2014)
Abstract
Identification of CD4+Foxp3+ Tregs and Th17 modified the historical Th1–Th2 paradigm. Currently, the Th17–Tregs dichotomy provides a dominant conceptual framework for the comprehension of immunity/inflammation and tolerance/immunosuppression in an increasing number of diseases. Targeting proinflammatory Th17 cells or immunosuppressive Tregs has been widely considered as a promising therapeutic strategy in the treatment of major human diseases, including autoimmunity and cancer. The efficacy and safety of such therapy rely on a thorough understanding of immunobiology and interaction of these two subsets of Th cells. In this article, we review recent progress concerning complicated interplay of Th17 cells and Tregs. There is compelling evidence that Tregs potently inhibit Th1 and Th2 responses; however, the inhibitory effect of Tregs on Th17 responses is a controversial subject. There is increasing evidence showing that Tregs actually promote the differentiation of Th17 cells in vitro and in vivo and consequently, enhanced the functional consequences of Th17 cells, including the protective effect in host defense, as well as detrimental effect in inflammation and in the support of tumor growth. On the other hand, Th17 cells were also the most potent Th subset in the stimulation and support of expansion and phenotypic stability of Tregs in vivo. These results indicate that these two subsets of Th cells reciprocally stimulate each other. This bidirectional crosstalk is largely dependent on the TNF–TNFR2 pathway. These mutual stimulatory effects should be considered in devising future Th17 cell- and Treg-targeting therapy.
Keywords: Foxp3, IL-17, TNF, TNFR2, immunosuppression, inflammation
Th17 signalling and downstream pathways:
A Case for Targeting Th17 Cells and IL-17A in SARS-CoV-2 Infections (2020)
Abstract
SARS-CoV-2, the virus causing COVID-19, has infected millions and has caused hundreds of thousands of fatalities. Risk factors for critical illness from SARS-CoV-2 infection include male gender, obesity, diabetes, and age >65. The mechanisms underlying the susceptibility to critical illness are poorly understood. Of interest, these comorbidities have previously been associated with increased signaling of Th17 cells. Th17 cells secrete IL-17A and are important for clearing extracellular pathogens, but inappropriate signaling has been linked to acute respiratory distress syndrome. Currently there are few treatment options for SARS-CoV-2 infections. This review describes evidence linking risk factors for critical illness in COVID-19 with increased Th17 cell activation and IL-17 signaling that may lead to increased likelihood for lung injury and respiratory failure. These findings provide a basis for testing the potential use of therapies directed at modulation of Th17 cells and IL-17A signaling in the treatment of COVID-19.
Th17 and the inflammatory response
Th17 cells differentiate in the setting of a proinflammatory cytokine milieu and secrete several proinflammatory cytokines, including IL-17A, IL-17F, and IL-22 (19). In mice, TGF-β and IL-6 increase Th17 cell differentiation via activation of the transcription factor STAT3 (32). In humans, the exact combination of cytokines necessary for initiation of Th17 cell differentiation is not established, but IL-1β and IL-23 both contribute to Th17 cell differentiation with possible contributions from TGF-β and IL-6 (19, 32) (Fig. 1). STAT3 activates transcription of retinoic acid receptor–related orphan receptor γt (RORγt). RORγt, also known as the “master regulator” of Th17 cell differentiation, activates transcription of genes such as IL-17A, IL-17F, and IL-22 (19). In humans, TGF-β has dose-dependent effects on T cell differentiation. At low levels, TGF-β increases RORγt transcription, but at higher levels, TGF-β promotes transcription of FOXP3, a regulator of differentiation of Tregs, leading to inhibition of RORγt transcription (19) and suppression of Th17 cell differentiation. Studies suggest that there is inhibition between different Th cell pathways as Th1 cytokines (IFN-γ) and Th2 cytokines (IL-4 and IL-12) are able to inhibit Th17 cell differentiation (32, 33) and TGF-β inhibits differentiation of Th1 and Th2 cells (33).
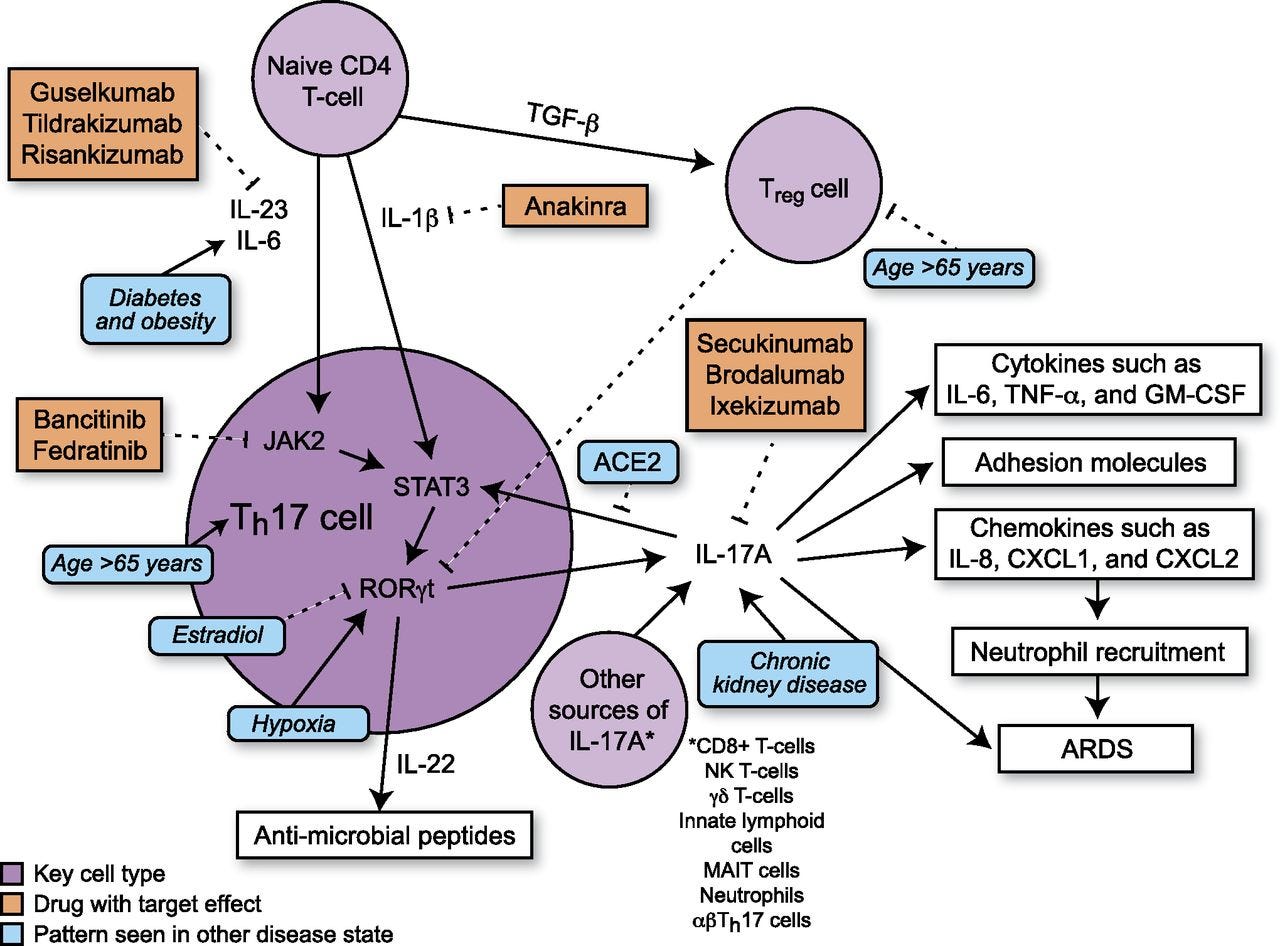
FIGURE 1.
Schematic showing Th17 differentiation and downstream signaling. Comorbidities of critical illness in COVID19 and their effects on Th17 signaling are depicted in blue. Inhibitors of Th17 signaling are depicted in orange.
Full paper:
https://www.jimmunol.org/content/205/4/892
TNF-α impairs differentiation and function of TGF-β-induced Treg cells in autoimmune diseases through Akt and Smad3 signaling pathway (2014)
Abstract
Deficiency in the TGF-β-induced regulatory T (iTreg) cell differentiation is associated with compromised immune homeostasis and plays a key role in many autoimmune diseases. Therapeutic intervention to enhance in situ iTreg differentiation has become a promising treatment modality for autoimmune diseases. Here we describe that the development of autoimmune inflammation in experimental autoimmune encephalomyelitis (EAE) is associated with selective impairment of iTreg differentiation largely due to the increased production of TNF-α. The neutralization of TNF-α markedly increases iTreg differentiation, leading to the amelioration of EAE, whereas the depletion of iTreg cells abolishes the therapeutic effect of an anti-TNF-α antibody. The inhibition of iTreg differentiation by TNF-α is mediated through a signaling cascade involving the induction of TNF receptor II (TNFR2) expression and the activation of Akt. The activated Akt in turn interacts with Smad3, resulting in the inhibition of TGF-β-induced Smad3 phosphorylation and consequently the reduction of p-Smad3 results in the decreased binding to the specific binding site of the foxp3 promoter, and finally foxp3 transcription itself. Interestingly, this regulatory pathway is iTreg cell specific as TNF-α does not activate Akt in naturally occurring regulatory T cells, therefore conferring a selective effect of TNF-α and its antagonism on iTreg cells. The study sheds new light on the critical role and underlying mechanism of TNF-α in the regulation of iTreg differentiation and provides a novel rationale for TNF-α antagonistic therapy for autoimmune diseases.
https://pubmed.ncbi.nlm.nih.gov/23243069/
Spike protein and pathophysiological pathways:
Comprehensive investigations revealed consistent pathophysiological alterations after vaccination with COVID-19 vaccines (2021)
Abstract
Large-scale COVID-19 vaccinations are currently underway in many countries in response to the COVID-19 pandemic. Here, we report, besides generation of neutralizing antibodies, consistent alterations in hemoglobin A1c, serum sodium and potassium levels, coagulation profiles, and renal functions in healthy volunteers after vaccination with an inactivated SARS-CoV-2 vaccine. Similar changes had also been reported in COVID-19 patients, suggesting that vaccination mimicked an infection. Single-cell mRNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) before and 28 days after the first inoculation also revealed consistent alterations in gene expression of many different immune cell types. Reduction of CD8+ T cells and increase in classic monocyte contents were exemplary. Moreover, scRNA-seq revealed increased NF-κB signaling and reduced type I interferon responses, which were confirmed by biological assays and also had been reported to occur after SARS-CoV-2 infection with aggravating symptoms. Altogether, our study recommends additional caution when vaccinating people with pre-existing clinical conditions, including diabetes, electrolyte imbalances, renal dysfunction, and coagulation disorders.
Reduction of CD8+ T cells and increase in classic monocyte contents were exemplary. Moreover, scRNA-seq revealed increased NF-κB signaling and reduced type I interferon responses, which were confirmed by biological assays and also had been reported to occur after SARS-CoV-2 infection with aggravating symptoms. Altogether, our study recommends additional caution when vaccinating people with pre-existing clinical conditions, including diabetes, electrolyte imbalances, renal dysfunction, and coagulation disorders.
Full paper:
https://www.nature.com/articles/s41421-021-00329-3
SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells (2020)
Abstract
Cytokine storm is suggested as one of the major pathological characteristics of SARS-CoV-2 infection, although the mechanism for initiation of a hyper-inflammatory response, and multi-organ damage from viral infection is poorly understood. In this virus-cell interaction study, we observed that SARS-CoV-2 infection or viral spike protein expression alone inhibited angiotensin converting enzyme-2 (ACE2) receptor protein expression. The spike protein promoted an angiotensin II type 1 receptor (AT1) mediated signaling cascade, induced the transcriptional regulatory molecules NF-κB and AP-1/c-Fos via MAPK activation, and increased IL-6 release. SARS-CoV-2 infected patient sera contained elevated levels of IL-6 and soluble IL-6R. Up-regulated AT1 receptor signaling also influenced the release of extracellular soluble IL-6R by the induction of the ADAM-17 protease. Use of the AT1 receptor antagonist, Candesartan cilexetil, resulted in down-regulation of IL-6/soluble IL-6R release in spike expressing cells. Phosphorylation of STAT3 at the Tyr705 residue plays an important role as a transcriptional inducer for SOCS3 and MCP-1 expression. Further study indicated that inhibition of STAT3 Tyr705 phosphorylation in SARS-CoV-2 infected and viral spike protein expressing epithelial cells did not induce SOCS3 and MCP-1 expression. Introduction of culture supernatant from SARS-CoV-2 spike expressing cells on a model human liver endothelial Cell line (TMNK-1), where transmembrane IL-6R is poorly expressed, resulted in the induction of STAT3 Tyr705 phosphorylation as well as MCP-1 expression. In conclusion, our results indicated that the presence of SARS-CoV-2 spike protein in epithelial cells promotes IL-6 trans-signaling by activation of the AT1 axis to initiate coordination of a hyper-inflammatory response.
https://pubmed.ncbi.nlm.nih.gov/33284859/
Pathological outcomes:
With tumors IL-17 is context sensitive with regard to pro or anti-tumor activity. What this paper appears to be saying is that if you already have a tumor then IL-17 can act to improve its blood supply and therefore rate of expansion, invasiveness and eventually metastasis, ie it may be pro-tumor to anything pre-existing.
In contrast, if you are immune competent and cancer free it can help promote anti-tumor inflammatory responses:
TH17 cells in tumour immunity and immunotherapy (2011)
Abstract
T helper 17 (TH17) cells have well-described roles in autoimmune disease. Recent evidence suggests that this effector T cell subset is also involved in tumour immunology and may be a target for cancer therapy. In this Review, we summarize recent findings regarding the nature and relevance of TH17 cells in mouse models of cancer and human disease. We describe the interplay between TH17 cells and other immune cells in the tumour microenvironment, and we assess both the potential antitumorigenic and pro-tumorigenic activities of TH17 cells and their associated cytokines. Understanding the nature of TH17 cell responses in the tumour microenvironment will be important for the design of more efficacious cancer immunotherapies.
Although the link between inflammation and cancer has been noted for more than a century, investigators have only recently started to address the cellular, molecular and genetic causal relationships between these two events. Compelling evidence has shown that inflammation orchestrates the microenvironment around tumours, contributing to the proliferation, migration and survival of cancer cells that can result in tumour invasion, migration and metastasis. However, inflammatory reactions in the tumour microenvironment are an important component of the tumour-associated immune response. Inflammatory cells and molecules may have crucial roles in initiating and maintaining protective antitumour immunity. The specific nature of the inflammatory response and the tissue context may determine the beneficial versus the detrimental effects of inflammation on tumour pathology.
T helper 17 (TH17) cells are an important inflammatory component and have been shown to promote inflammation in a number of autoimmune diseases1–8. Research on these cells is rapidly evolving, and recent reviews have extensively covered basic TH17 cell biology, TH17 cell lineage development and the relevance of TH17 cells in autoimmune diseases 1–4,9,10. In this Review, we summarize recent reports of TH17 cells in patients with cancer and in mouse tumour models. We examine the phenotype, recruitment, generation and function of tumour-associated TH17 cells, focusing on their production of cytokines and on their interplay with other immune cells in the tumour microenvironment. Finally, we discuss the clinical relevance of TH17 cells in tumour immunology and highlight their therapeutic potential.
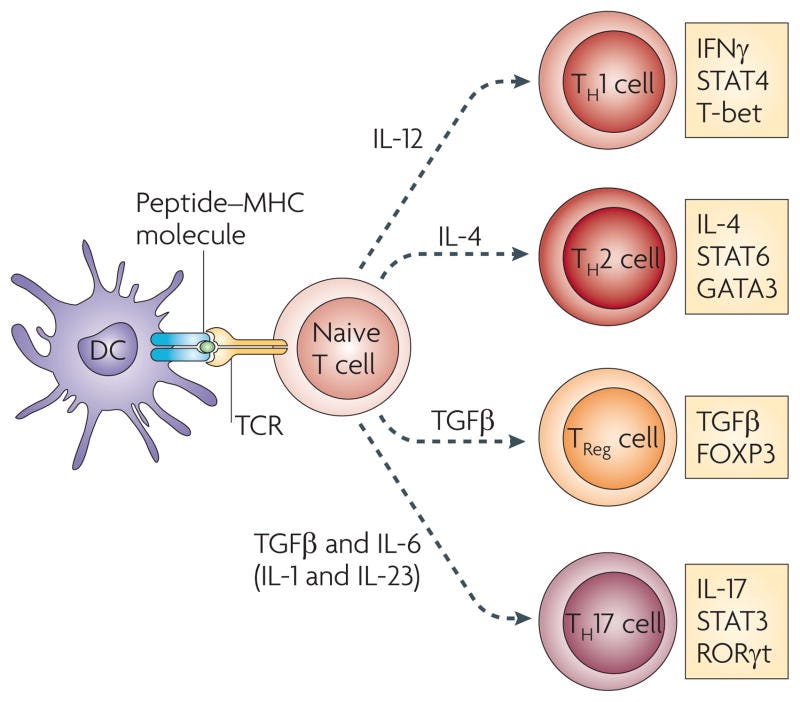
Differentiation of helper T cell subsets
Following activation by antigen-presenting cells such as dendritic cells (DCs), naive CD4+ T cells can be polarized into different effector T cell subsets — T helper 1 (TH1), TH2, TH17 and regulatory T (TReg) cells — depending on the local cytokine environment. The differentiation of each of these effector T cell subsets is controlled by distinct sets of transcription factors. In the presence of interleukin-6 (IL-6) and transforming growth factor-β (TGFβ), naive T cells can differentiate into TH17 cells, which are characterized by expression of the transcription factors retinoic acid receptor-related orphan receptor-γt (RORγt) and signal transducer and activator of transcription 3 (STAT3). Furthermore, IL-1 and IL-23 can promote and/or stabilize TH17 cell differentiation and expansion. FOXP3, forkhead box P3; GATA3, GATA-binding protein 3; IFNγ, interferon-γ; TCR, T cell receptor.
TH17 cells and antitumour immunity
Table 2:
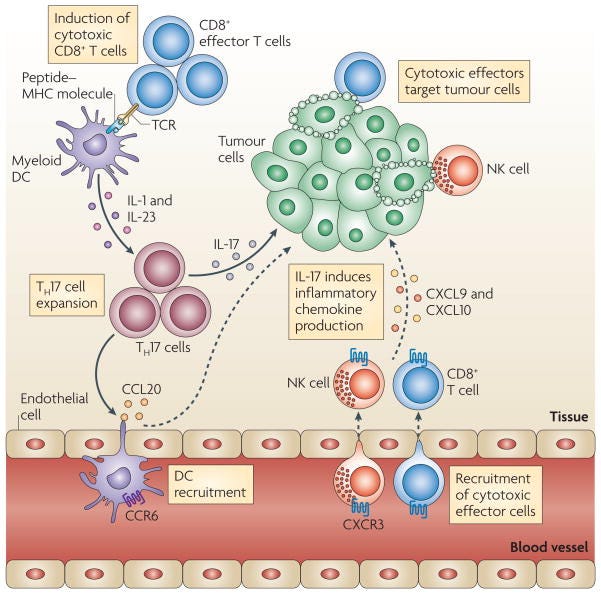
TH17 cells and antitumour immunity
T helper 17 (TH17) cells traffic to the tumour microenvironment and are expanded by antigen-presenting cells, such as myeloid dendritic cells (DCs) through interleukin-1 (IL-1) and IL-23. TH17 cells promote the trafficking and retention of effector T cells and natural killer (NK) cells in the tumour environment, through inducing the production of the chemokines CXC-chemokine ligand 9 (CXCL9) and CXCL10 by primary tumour cells. In addition, TH17 cells induce the production of CC-chemokine ligand 20 (CCL20) by tumour cells and this leads to the recruitment of CC-chemokine receptor 6 (CCR6)+ DCs. Therefore TH17 cells can promote protective antitumour immunity by inducing the recruitment of pro-inflammatory immune effector cells; TCR, T cell receptor.
Pro-tumour role of TH17 cell-associated cytokines
IL-17 and TH17 cells
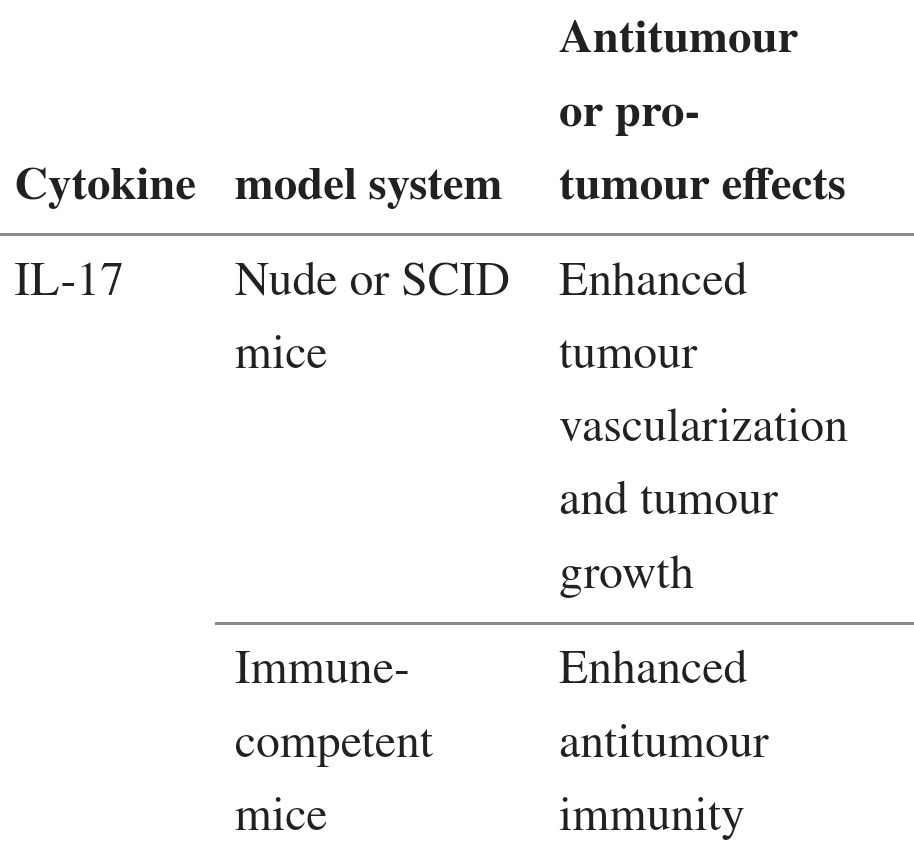
Although IL-17 is the signature cytokine of TH17 cells, the production of IL-17 is not the sole function of TH17 cells. Thus, the biological activities of IL-17 should not be equated with the biological activities of TH17 cells (BOX 1). In addition to leukocytes of the immune system, the cellular targets of IL-17 in the tumour microenvironment can be vascular endothelial cells, stromal cells and cells of the tumour itself. Early studies showed that exogenous IL-17 could promote tumour growth by inducing tumour vascularization, particularly in immune-deficient nude mice and severe combined immunodeficient (SCID) mice71–73 (TABLE 2). However, the overall effect of IL-17 on tumour development and growth might be different in immune-competent hosts, as shown by the potent antitumour effects mediated by IL-17 in immune-competent mice60,61 (TABLE 2). Furthermore, as a result of differences in local concentrations, bioavailability and potential targets, the biological activities of endogenous IL-17 (such as IL-17 derived from TH17 cells) and exogenously administered IL-17 might differ. Two recent reports have shown that, in mice, endogenous IL-17 promotes Bacteroides fragilis-induced tumour formation74 and tumour growth in a transplanted tumour model75. IL-17 induces IL-6 production by tumour cells and tumour-associated stromal cells, which in turn activates STAT3, an oncogenic transcription factor that upregulates pro-survival and pro-angiogenic genes75. Furthermore, although IL-17 deficiency leads to increased numbers of IFNγ-producing NK cells in the tumour-draining lymph nodes of tumour-bearing mice59, it has also been reported that IL-17 can decrease NK cell activity in a mouse model of dermatitis76. Therefore IL-17 can promote tumour growth in certain tumour-bearing mouse models, and the effects of IL-17 on tumour growth might be highly context dependent (BOX 1; TABLE 2).
Full paper:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3242804/#__ffn_sectitle
IL-17 appears to be the early onset trigger for several autoimmune inflammatory disorders. The Th17 cell types can change later though so a treatment that may work at an early stage will not work once a condition is more advanced. Its very complex!
The pathogenicity of Th17 cells in autoimmune diseases (2019)
Abstract
IL-17-producing T helper (Th17) cells have been implicated in the pathogenesis of many inflammatory and autoimmune diseases. Targeting the effector cytokines IL-17 and GM-CSF secreted by autoimmune Th17 cells has been shown to be effective for the treatment of the diseases. Understanding a molecular basis of Th17 differentiation and effector functions is therefore critical for the regulation of the pathogenicity of tissue Th17 cells in chronic inflammation. Here, we discuss the roles of proinflammatory cytokines and environmental stimuli in the control of Th17 differentiation and chronic tissue inflammation by pathogenic Th17 cells in humans and in mouse models of autoimmune diseases. We also highlight recent advances in the regulation of pathogenic Th17 cells by gut microbiota and immunometabolism in autoimmune arthritis.
Keywords: Autoimmune arthritis; EAE; GM-CSF; IL-17; Rheumatoid arthritis; Th17 cells.
Introduction
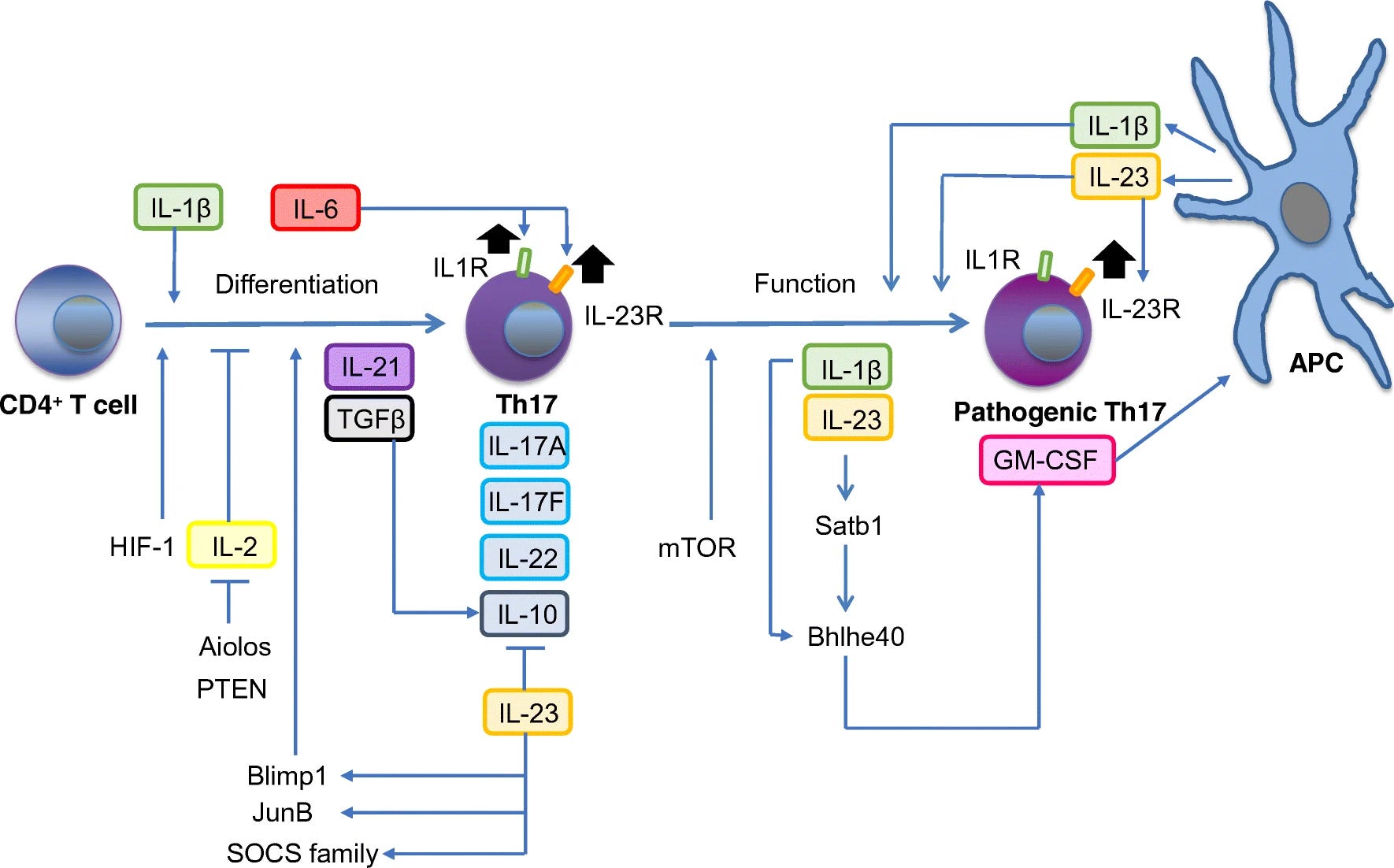
CD4+ T helper (Th) cells play pivotal roles in the tissue destruction of many inflammatory and autoimmune diseases such as multiple sclerosis (MS), rheumatoid arthritis (RA), psoriasis, and inflammatory bowel diseases [1]. Among Th subsets, interleukin-17 (IL-17)-producing Th (Th17) cells are classified as an inflammatory Th subset, resulting in chronic tissue inflammation and subsequent organ failure [2, 3]. Indeed, some biologic agents targeting the effector cytokines of Th17 cells have been approved for the treatment of certain immune-mediated diseases and we will see the expansion of diseases, which could be treatable by them. Therefore, understanding the differentiation and pathogenic functions of Th17 cells is crucial for the development of a novel immunotherapy for Th17-associated inflammatory diseases. Here, we focus on how Th17 cells acquire the terminal effector function, in particular, the pathogenicity in disease models and how environmental cues modulate Th17 cells that orchestrate chronic tissue inflammation in SKG mice, an animal model of RA. Finally, we discuss the role of gut microbiota and immunometabolism in modulating Th17 cells of patients with RA.
The regulation of Th17 differentiation and effector function
…
Human Th17 cells and their role in neuroinflammation
Similar to mouse Th17 cells, the differentiation of human Th17 cells in vitro requires IL-1, IL-6, IL-23, and TGFβ. Several studies initially demonstrated that IL-1β, IL-6, and IL-23 but not TGFβ were sufficient to induce the differentiation of human Th17 cells [60, 61]. However, careful assessments later reconciled the role of TGFβ in human Th17 differentiation, showing that TGFβ, IL-23, and IL-1β (or IL-6) under serum-free conditions were essential in driving Th17 differentiation because culture medium contained serum-derived TGFβ or AhR ligands [62, 63]. Consistent with mouse Th17 cells, IL-23 plays the major role in human Th17 differentiation as human naïve CD4+ T cells can immediately respond to IL-23 and the IL-23R expression is further upregulated by IL-23 signals in the presence of additional IL-1β [62, 64]. Furthermore, dominant-negative mutations in STAT3, a key transcription factor downstream of IL-6 and IL-23 signaling, were responsible for disease manifestations of hyper-immunoglobulin E syndrome and impaired IL-17 production and the differentiation of Th17 cells, supporting roles of STAT3 in IL-6 and IL-23 signaling pathways and Th17 differentiation in humans [65, 66].
Before the discovery of Th17 subset, IL-17 was identified as the highest-ranking gene expressed in the CNS of MS patients [67]. It is of note that accumulation of Th17 cells were the first wave of T cells infiltrating the CNS [68], followed by the infiltration of DCs, macrophages, and other cells, which further promote and sustain tissue inflammation. In addition, a number of studies have shown that a single nucleotide polymorphism (SNP) in IL-23R is linked to a number of human autoimmune diseases [1], indicating that IL-23 signaling is essential for evoking a pathogenic feature in Th17 cells. IL-23 is also associated with the risk of MS [69, 70]. Since GM-CSF have a pivotal role in Th17 cells for encephalitogenicity in mice [41, 52], the findings that there were elevated levels of GM-CSF in the cerebrospinal fluid and serum of active MS patients with relapsing-remitting type and increased GM-CSF production of T cell from the peripheral blood and brain lesion of MS suggest a crucial role of GM-CSF in disease manifestations and tissue inflammation.
The pathogenicity of Th17 cells in autoimmune arthritis models
RA is a systemic autoimmune disease that affects about 1% of general population worldwide, characterized by chronic joint inflammation and bone destruction [73]. Although the exact pathogenesis remains to be determined, it has been recognized that Th subsets play an important role in RA pathogenesis based on human leukocyte antigen (HLA)-DRB1 identified as the strongest disease risk gene, abundant T cells and macrophages infiltrating into synovial membrane in RA patients, and presence of circulating autoantibody such as rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPA) [73,74,75,76]. When the Th1/Th2 paradigm dominated in the pathogenesis of autoimmune diseases before the discovery of Th17 cells, RA as well as MS was previously thought to be Th1-mediated diseases. However, the levels of Th-1-mediated cytokines such as IFN-γ in RA synovium were relatively low compared with those of TNF-α, IL-1, or IL-6 derived from inflammatory macrophage- and fibroblast-like synoviocytes (FLS) [77, 78]. The importance of these macrophage- and FLS-derived proinflammatory cytokines in RA is evident based on the efficacy of anti-cytokine therapy, such as anti-TNF or anti-IL-6 therapy, which brought a paradigm shift in RA treatment.
The discovery of Th17 cells have shed new insights into how inflammatory Th subsets contribute to the initiation of RA and form a proinflammatory cytokine network, leading to chronic inflammation, in particular, in animal models of autoimmune arthritis. There are several murine models to understand the pathogenesis of RA. Firstly, collagen-induced arthritis (CIA), which is induced by immunizing mice with type II collagen and whose pathogenicity is totally dependent on the generation of anti-collagen autoantibodies, is one of the well-established arthritis models. K/BxN mice, another model of arthritis, develop spontaneous arthritis mediated by the arthritogenic anti-glucose-6-phosphate isomerase (GPI) autoantibody [79, 80]. It has been demonstrated that IL-17 plays a pathogenic role to a greater or lesser extent in these murine models. However, with regard to T cell-dependent, but not autoantibody-dependent autoimmune arthritis, SKG mice are a suitable model to focus how T cells mediate autoimmune arthritis.
We have proposed a possible mechanism of how self-reactive (arthritogenic) Th cells become effector Th17 cells, migrate to synovium and initiate joint inflammation in SKG mice. Self-reactive T cells become activated in the periphery via recognition of class II MHC/self-peptide complexes expressed by APCs, and stimulate APCs through CD40/CD40L interaction to upregulate CD80/CD86, which further activate these T cells to proliferate. Activated APCs secrete a large amount of IL-6 (also IL-1, IL-23, and TNFα), together with surrounding tissue-derived TGF-β, that induces the differentiation of effector Th17 cells. Expanded arthritogenic Th17 cells predominantly express CCR6 and migrate to joints in response to CCL20, the ligand of CCR6, which is secreted by activated FLSs. Indeed, treatment with anti-CCR6 blocking mAb significantly inhibits the infiltration of Th17 cells into joints and reduces the severity of arthritis in SKG mice. In vitro, CCL20 expression in synoviocytes is promoted by IL-17, IL-1β, or TNFα, whereas IFN-γ or IL-4 inhibits its expression. Thus, once arthritogenic Th17 cells are activated and recruited into joints to initiate inflammation, synoviocytes further recruit Th17 cells in a feed-forward mechanism by which CCL20 production is augmented by proinflammatory cytokines such as IL-17, IL-1β, or TNFα derived from both activated synoviocytes and Th17 cells (Fig. 2). Expression of CCR6 in Th17 cells and CCL20 in synoviocytes are also observed in RA patients with a significant correlation between the amounts of IL-17 and CCL20 in RA joints [90]. Furthermore, recent genome-wide association study (GWAS) studies identified CCR6 as a disease susceptibility gene of RA, which together implies the pathogenicity of Th17 in RA and a shared mechanism of Th17 recruitment in inflamed joints.

A possible mechanism of self-reactive Th17 differentiation in SKG mice

Th17 cells orchestrate chronic joint inflammation in SKG mice
Human Th17 cells in rheumatoid arthritis
As we have discussed above, there are several observations that support the pathogenesis of Th cells in RA. The efficacy of CTLA4-Ig treatment in RA indicates a central role of Th cells. In addition, recent GWAS study further identified RA risk loci that are linked to T cell function (e.g., PTPN22, CD28, CD40), and are even more specific to Th17-associated molecules such as CCR6 [91, 96,97,98,99,100]. However, the role of Th17 cells in RA is not as clear as animal models yet. Many reports agree that levels of IL-17 are increased in synovial fluid (SF) and synovial tissue (ST) in RA [101,102,103,104]. Kirkham et al. reported that the levels of IL-17 expression in ST are predictive of joint damage in RA patients, which could be the result of enhanced osteoclasts activation mediated by IL-17-rich environment [105]. However, it is controversial whether Th17 cells increase in the peripheral blood, SF, or ST in RA [106,107,108,109,110]. Furthermore, it was reported in ST that the vast majority of IL-17-secreting cells were not Th17 cells, which is one of the major differences from animal models of arthritis. The exact sources of IL-17 in ST remain unclear [101, 111]. Hueber et al. reported that the major source of IL-17 was synovial mast cells, which promote IL-17 production in stimuli by TNFα, IgG complexes or C5a, whereas Kan et al. later reported that the number of IL-17-producing mast cells and the frequency of IL-17-producing mast cells among all the IL-17+ cells in ST were comparable between RA and osteoarthritis (OA) patients, raising a question to the contribution of synovial mast cells as a source of IL-17 [111, 112]. Furthermore, the frequencies of Th1 cells among CD4+ T cells in SF and ST of RA patients are much larger than those of Th17 cells [81, 110]. However, the plasticity of Th17 cells toward Th1-like cells may be able to give a possible explanation for this observation. Nistala et al. showed the presence of IFN-γ-producing Th17 cells (hereafter Th17/Th1) in SF from patients with juvenile idiopathic arthritis (JIA), which express both Th1 and Th17 transcription factors such as T bet and RORC2 [113]. They also demonstrated in vitro that Th17 cells can be converted to Th17/Th1-cell phenotype in IL-12high TGFβlow conditions that resembles the SF environment of JIA. Furthermore, TCRβ (TRBV) repertoire analysis revealed that synovial Th17/Th1 cells share TCRβ repertoire oligoclonality with Th17 cells, indicating that Th17/Th1 cells may be deviated from Th17 cells. Moreover, CD161, one of the surface marker of Th17 cells in human as well as CCR6, is expressed not only in Th17/Th1 cells, but also in a number of Th1 cells in SF. These findings together suggest that a certain subpopulation of IFN-γ-producing CD4+ T cells in arthritic joints may originate from Th17 cells, which shift to Th1-like phenotype via an intermediate state of Th17/Th1 cells, when they encounter a IL-12 high TGFβ low environmental cue in the local inflammatory site. In fact, there are accumulating evidence that IFN-γ+ ex-Th17 cells are enriched in SF of JIA and RA in comparison to peripheral blood [113,114,115]. In addition, recent studies revealed that approximately 80% of GM-CSF-producing CD4+ T cells in joints of JIA and RA co-express IFN-γ, but rarely express IL-17 [116, 117]. The majority of these IFN-γ+ GM-CSF+ CD4+ T cells express CD161, indicating that the subpopulation of IFN-γ+ ex-Th17 cells also actively produce GM-CSF [117].
As Th17 cells have been highlighted in the pathogenesis of RA, IL-17 inhibitors as a new potential biologic agent were trialed for RA treatment. In phase II trials, treatment with anti-IL-17 antibodies, secukinumab or ixekizumab, have demonstrated preliminary efficacy in RA with biologic-naïve or who failed to respond to TNF inhibitors or methotrexate [118,119,120,121]. However, in phase III trials, IL-17 inhibition showed no incremental benefit over the biologic agents currently approved to non-responders for TNF inhibitors [122, 123]. Although there is no clinical trial that has focused on evaluating the efficacy of IL-17 inhibitors in the onset or early stage of RA, these results indicate that IL-17 inhibitors alone are not sufficient enough to suppress ongoing chronic inflammation in established RA. Notably, it has been reported that higher levels of IL-17 in SF exist at the early stage of RA compared with the established stage and those of IL-17 in sera rather decreases after the onset of RA [124, 125]. Taking these findings together, Th17 cells may have different roles at different phases of RA. Th17 cells initiate joint inflammation via IL-17 by stimulating FLSs, promoting osteoclast differentiation and recruiting abundant neutrophils and more Th17 cells. However, soon after the onset of inflammation, a number of Th17 cells may become IFN-γ+ ex-Th17 cells in response to surrounding cytokine environments in the arthritic joints and simultaneously begin to actively secrete GM-CSF together with other GM-CSF-producing cells such as activated FLSs and synovial ILCs that synergistically provoke synovial macrophages to secrete a large amount of tissue destructive molecules and proinflammatory cytokines such as TNF-α or IL-6, leading to chronic inflammation. Thus, the pathogenesis of Th17 cells in RA may shift from “IL-17-producer,” as an initiator of the disease, into “GM-CSF-producer,” as an organizer of chronic inflammation. GM-CSF has been highlighted as a key mediator in RA based on recent clinical trials using GM-CSF inhibitors for RA patients [126,127,128,129]. From our point of view, IL-17 inhibitors may show a better clinical outcome for early onset RA patients, although it is not easy to identify and test such patients in a clinical trial.
Psoriasis, psoriatic arthritis (PsA), and ankylosing arthritis (AS) have been implicated in Th17-mediated autoimmune diseases in humans. Unlike RA, IL-17 inhibition shows marked clinical efficacies in psoriasis or PsA patients (also IL-12/23p40 inhibition) and AS patients, indicating the proof of concept in the pathogenesis of IL-17 in these diseases [130,131,132,133,134]. Further studies will be required to understand how IL-17-type/disease-specific tissue inflammation changes before and after the treatment with IL-17 inhibitors.
Full paper:
https://pubmed.ncbi.nlm.nih.gov/30891627/
A therapeutic!
Plus see:
Effects of resveratrol on Th17 cell-related immune responses under tacrolimus-based immunosuppression (2019)
Abstract
Background
We previously reported that tacrolimus (Tac) does not decrease T helper 17 cells (Th17) response in kidney transplantation. In this study, we evaluated whether Resveratrol (Resv) has immunosuppressive effects by decreasing Th17 responses in Tac-based immunosuppression.
Methods
We investigated the effects of Resv under Tac-treatment conditions, on CD4+ T cell differentiation to Th17 cells in peripheral blood mononuclear cells (PBMCs), and proliferation of CD4+ T cells co-cultured with human renal proximal tubular epithelial cells (HRPTEpiCs). The effects of Resv on Th17 cells were tested in the murine skin transplant model.
Results
In PBMCs, Tac did not but combination of Tac and Resv further suppressed Th17 immune response. In the co-culture study, combination of Resv to Tac significantly decreased HRPTEpiC-induced T cell proliferation compared to Tac alone. Resv treatment in the Jurkat cell induced the expression of AMP-activated protein kinase and suppressed the expression of mammalian target of rapamycin (mTOR), suggesting blocking Th17 pathway by Resv. In the murine skin transplant model, combination of Resv to Tac significantly prolonged skin graft survival accompanied by the suppression of Th17 cells, compared to either the Tac-alone or control groups.
Conclusion
The results of our study suggest that Resv provides additional immunosuppressive effects to Tac by suppressing effector CD4+ T cells, especially Th17 cells, in the transplantation setting.


Full paper:
https://bmccomplementmedtherapies.biomedcentral.com/articles/10.1186/s12906-019-2464-1

My name is Dr. Christina Parks and I am spearheading an alliance of practitioners looking at root causes of immune dysfunction. One practioner is seeing high rates of IL-10 and IL-17 production in long haulers as well as high INF-gamma. Clearly, you are a cytokine signaling person, as am I. I would love to connect and get your insight and reading recommendations. I don't want to leave my personal contact information on a public forum, but perhaps we can connect with FB messenger. https://www.facebook.com/christina.parks.752/
The work you are putting into this is hugely appreciated.
Thank you from someone who can follow about 10% of what you explained above.