Any extracts used in the following article are for non commercial research and educational purposes only and may be subject to copyright from their respective owners.
This Substack relates several pathological processes, from phenotypical presentation to the TLR2-dependent activation of the NF-κB pathway, how SARS-CoV-2 spike protein induces transcriptional upregulation of Snail, how SNAIL opposes miR-200, how miR-200b & c work to suppress inflammation and act as tumor suppressors. MicroRNAs and carcinogenesis are also explored.
Snail gets its name from zinc finger protein SNAI1.
Feedback mechanisms are involved in some of these processes, so pathway reversion may be involved and at times miR-200 can act as a tumor promoter.
The final point is that ACE2 depletion can lead to angiogenesis, which in itself can mediate tumorogenesis & metastasis, explored in a previous substack.
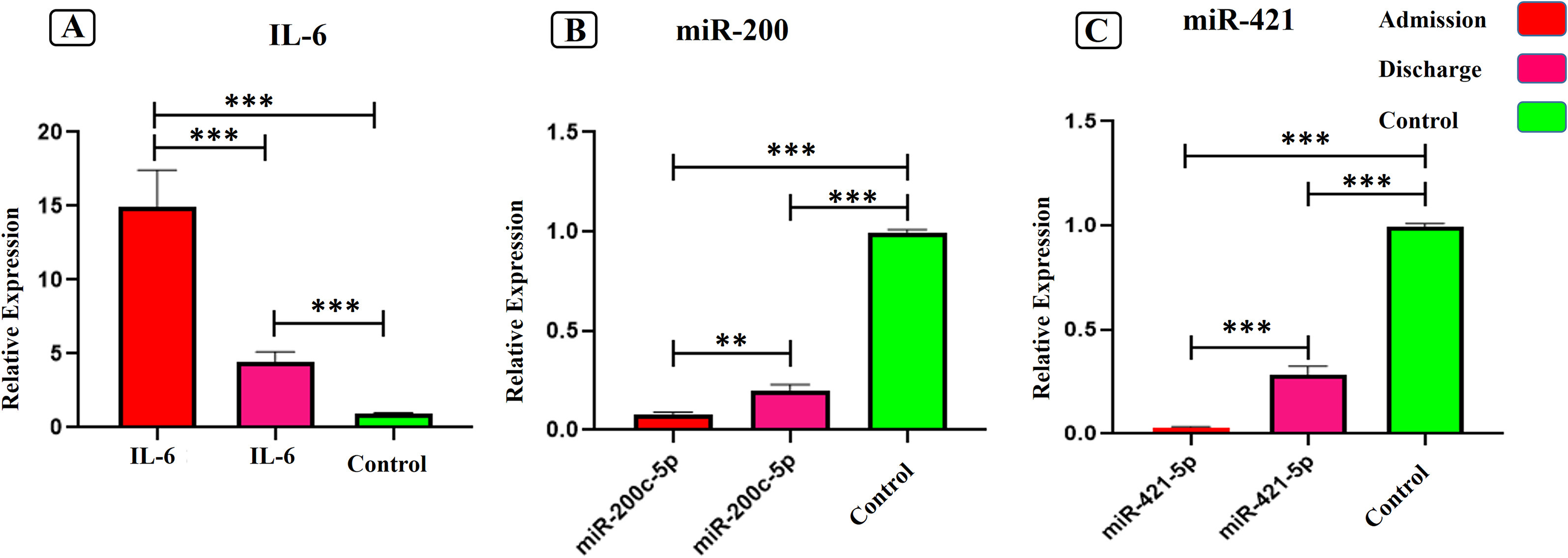
Evaluation of miR-200c-3p and miR-421-5p levels during immune responses in the admitted and recovered COVID-19 subjects (2021)
Angiotensin-converting enzyme 2 (ACE2) acts as a key receptor for the spike of SARS-CoV-2. Two main microRNAs (miRs), miR-200c-3p and miR-421-5p, are considered to modulate the expression of ACE2 gene and alterations in the expression of these miRNAs may influence the outcomes of COVID-19 infection. Accordingly, we examined whether miRNAs directing ACE2 expression altered in the SARS-CoV-2 infection. 30 patients with COVID-19 included in the study. At the time of admission and discharge, the expression of miR-200c-3p and miR-421-5p, inflammatory cytokine IL-6, and regulatory T cells' expression profiles (CD4, CD25, and Foxp3) were examined using quantitative real-time PCR method. At the time of admission, the expression levels of miR-200c-3p and miR-421-5p as well as CD4, CD25, and Foxp3 significantly decreased while IL-6 expression notably enhanced. However, by the time of discharge, the expression levels of the genes were opposite to the time of admission. Moreover, Pearson correlation analysis indicated that IL-6 expression negatively correlated with Foxp3 and miR-200c-3p expressions despite miR-421-5p and miR-200c-3p positively correlated at admission time. By manipulating miR-200c-3p and miR-421-5p expressions and controlling the ACE2 level, it is plausible to modulate the inflammation by reducing IL-6 and maintenance tolerance hemostasis during COVID-19 infection.
Keywords
COVID-19ACE-2miR-200c-3pmiR-421-5pIL-6
"Among various miRNAs subjected to ACE2 regulator (Nersisyan et al., 2020), the miR-200 family, especially hsa-miR-200c-3p directly targets 3’ UTR of ACE2 mRNA leading to decreased ACE2 expression. Expression of miR-200c-3p is induced through the NF-κB pathway during infection with the pandemic flu strain H5N1 and is associated with acute respiratory distress syndrome. In particular, the NF-κB signaling pathway is hyper-activated in SARS-CoV infections, suggesting that miR-200c-3p could also be up-regulated in patients with COVID-19, which subsequently result in decreased levels of ACE2 protein (Onabajo et al., 2020). Additionally, ACE2 is subjected to post-transcriptional regulation by miR-421-5p which is implicated in the development of thrombosis".
Pathways involved:
SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway
Abstract
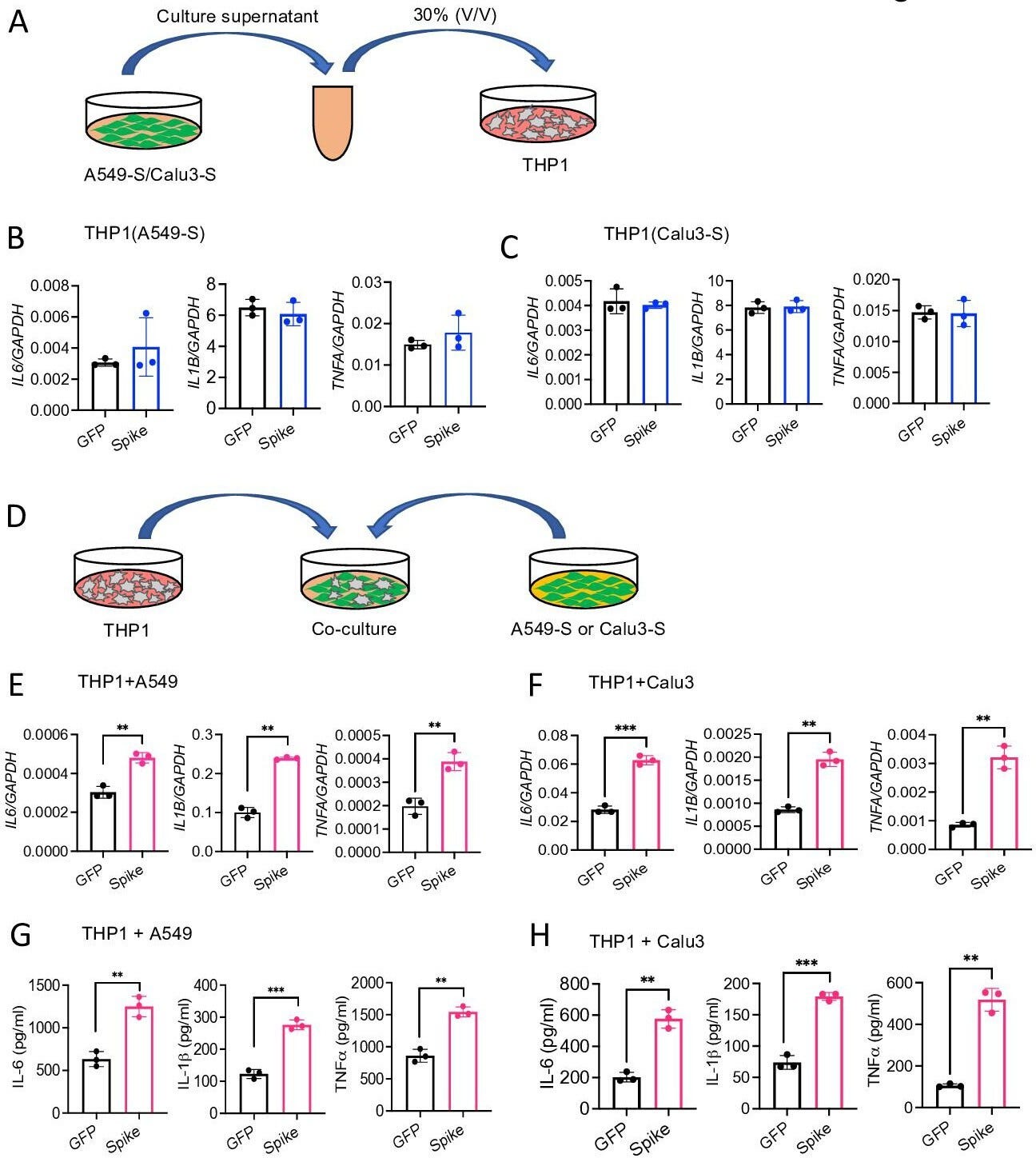
The pathogenesis of COVID-19 is associated with a hyperinflammatory response; however, the precise mechanism of SARS-CoV-2-induced inflammation is poorly understood. Here, we investigated direct inflammatory functions of major structural proteins of SARS-CoV-2. We observed that spike (S) protein potently induced inflammatory cytokines and chemokines, including IL-6, IL-1β, TNFα, CXCL1, CXCL2, and CCL2, but not IFNs in human and mouse macrophages. No such inflammatory response was observed in response to membrane (M), envelope (E), and nucleocapsid (N) proteins. When stimulated with extracellular S protein, human and mouse lung epithelial cells also produced inflammatory cytokines and chemokines. Interestingly, epithelial cells expressing S protein intracellularly were non-inflammatory, but elicited an inflammatory response in macrophages when co-cultured. Biochemical studies revealed that S protein triggers inflammation via activation of the NF-κB pathway in a MyD88-dependent manner. Further, such an activation of the NF-κB pathway was abrogated in Tlr2-deficient macrophages. Consistently, administration of S protein-induced IL-6, TNF-α, and IL-1β in wild-type, but not Tlr2-deficient mice. Notably, upon recognition of S protein, TLR2 dimerizes with TLR1 or TLR6 to activate the NF-κB pathway. Taken together, these data reveal a mechanism for the cytokine storm during SARS-CoV-2 infection and suggest that TLR2 could be a potential therapeutic target for COVID-19.
Lung epithelial cells produce inflammatory molecules in response to SARS-CoV-2 S protein.
(A, B) A549 or Calu3 cells were incubated with SARS-CoV-2 S1 or S2 proteins (500 ng/ml) for 12 and 24 hr. The expression of inflammatory cytokines and chemokines was measured by real-time RT-PCR. (C) Primary mouse lung epithelial cells were stimulated with S2 (500 ng/ml) for 12 and 24 hr. The expression of inflammatory cytokines and chemokines was measured by real-time RT-PCR. (D, E) Calu3 cells or mouse lung primary epithelial cells were stimulated with S2 (500 ng/ml). Culture supernatant collected at 12 and 24 hr were analyzed for IL-6, IL-1β, and TNFα by ELISA. *p<0.05, **p<0.001, ***p<0.0001, ****p<0.00001 by unpaired Student’s t-test. Experiments in (A, B) were repeated three times. Other experiments were repeated two times and data of representative experiments are presented.
Epithelial-mesenchymal transition induced by SARS-CoV-2 required transcriptional upregulation of Snail (2021)
Abstract
The engagement of human angiotensin-converting enzyme 2 (hACE2) and SARS-CoV-2 spike protein facilitate virus spread. Thus far, ACE2 and TMPRSS2 expression is correlated with the epithelial-mesenchymal transition (EMT) gene signature in lung cancer. However, the mechanism for SARS-CoV-2-induced EMT has not been thoroughly explored. Here, we showed that SARS-CoV-2 induces EMT phenotypic change and stemness in breast cancer cell model and subsequently identified Snail as a modulator for this regulation. The in-depth analysis identifies the spike protein (S), but not envelope (E), nucleocapsid (N), or membrane protein (M), of SARS-CoV-2 induces EMT marker changes. Suppression of Snail expression in these cells abrogates S protein-induced invasion, migration, stemness, and lung metastasis, suggesting that Snail is required for SARS-CoV-2-mediated aggressive phenotype in cancer. This study reveals an important oncogenic role of SARS-CoV-2 in triggering breast cancer metastasis through Snail upregulation.
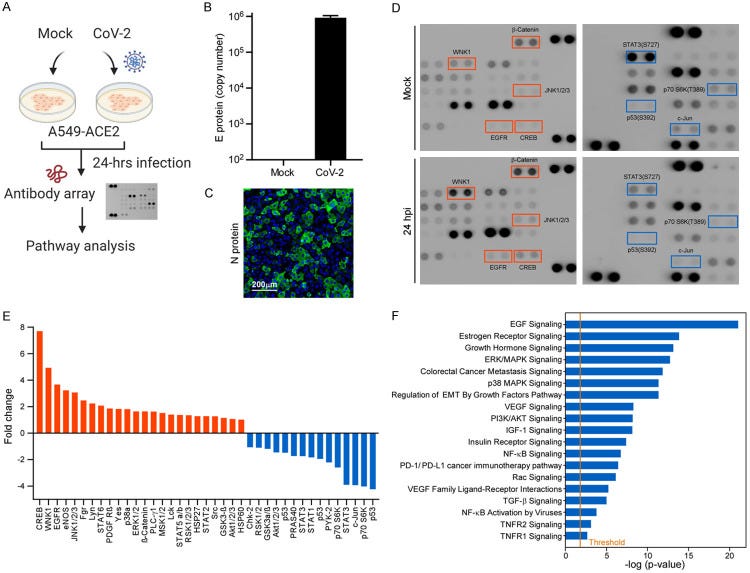
SARS-CoV-2 induces cancer cells to undergo EMT. A. Flowchart of the non-biased screening using a phospho-antibody array. B. Virus loading of SARS-CoV-2 in A549-ACE2 cells. Copy number of the viral genome was determined using a single strand DNA standard.
Figure 1
SARS-CoV-2 induces cancer cells to undergo EMT. A. Flowchart of the non-biased screening using a phospho-antibody array. B. Virus loading of SARS-CoV-2 in A549-ACE2 cells. Copy number of the viral genome was determined using a single strand DNA standard.
A candidate for Walters “oncogenic flash drive”:
"Suppression of Snail expression in these cells abrogates S protein-induced invasion, migration, stemness, and lung metastasis, suggesting that Snail is required for SARS-CoV-2-mediated aggressive phenotype in cancer. This study reveals an important oncogenic role of SARS-CoV-2 in triggering breast cancer metastasis through Snail upregulation."
"In-depth analysis of the relation between SARS-CoV-2 and cancer metastasis may help reduce COVID-19-mediated cancer aggressiveness."
"Several lines of evidence suggest that SARS-CoV-2-mediated Snail expression in breast cancer cells contributes to aggressive phenotype. First, we demonstrated that SARS-CoV-2 upregulates many oncogenic pathways, including EMT. Secondly, Ace2 expression is positively correlated with Snail expression in breast cancer cells. Thirdly, many canonical modules in NF-κB signaling were known to induce Snail expression [26]. Lastly, Snail expression is required for the SARS-CoV-2 or spike-mediated EMT changes. Although Snail contains a functional p65 binding motif on the promoter, we propose that SARS-CoV-2 induced breast cancer EMT is coordinately through canonical NF-κB signaling involved in Snail."
Snail and the microRNA-200 Family Act in Opposition to Regulate Epithelial-to-Mesenchymal Transition and Germ Layer Fate Restriction in Differentiating ESCs (2011)
Abstract
The reprogramming of somatic cells to inducible pluripotent stem cells requires a mesenchymal-to-epithelial transition. While differentiating ESCs can undergo the reverse process or epithelial-to-mesenchymal transition (EMT), little is known about the role of EMT in ESC differentiation and fate commitment. Here, we show that Snail homolog 1 (Snail) is expressed during ESC differentiation and is capable of inducing EMT on day 2 of ESC differentiation. Induction of EMT by Snail promotes mesoderm commitment while repressing markers of the primitive ectoderm and epiblast. Snail's impact on differentiation can be partly explained through its regulation of a number of ESC-associated microRNAs, including the microRNA-200 (miR-200) family. The miR-200 family is normally expressed in ESCs but is downregulated in a Wnt-dependent manner during EMT. Maintenance of miR-200 expression stalls differentiating ESCs at the epiblast-like stem cell (EpiSC) stage. Consistent with a role for activin in maintaining the EpiSC state, we find that inhibition of activin signaling decreases miR-200 expression and allows EMT to proceed with a bias toward neuroectoderm commitment. Furthermore, miR-200 requires activin to efficiently maintain cells at the epiblast stage. Together, these findings demonstrate that Snail and miR-200 act in opposition to regulate EMT and exit from the EpiSC stage toward induction of germ layer fates. By modulating expression levels of Snail, activin, and miR-200, we are able to control the order in which cells undergo EMT and transition out of the EpiSC state. Stem Cells 2011;29:764–776
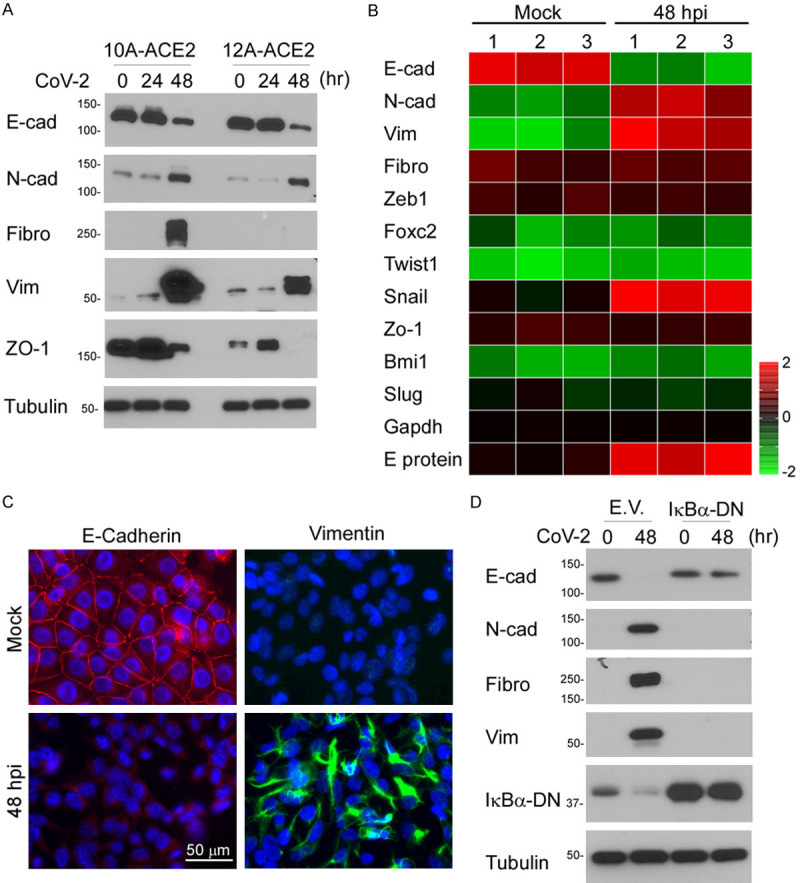
Snail promotes epithelial-to-mesenchymal transition in day 2 differentiating ESCs. (A): E-cadherin protein expression during MC50 ESC differentiation was analyzed by flow cytometry in serum-containing differentiation conditions without (black line) or with (gray line) addition of the Wnt inhibitor DKK at day 0.
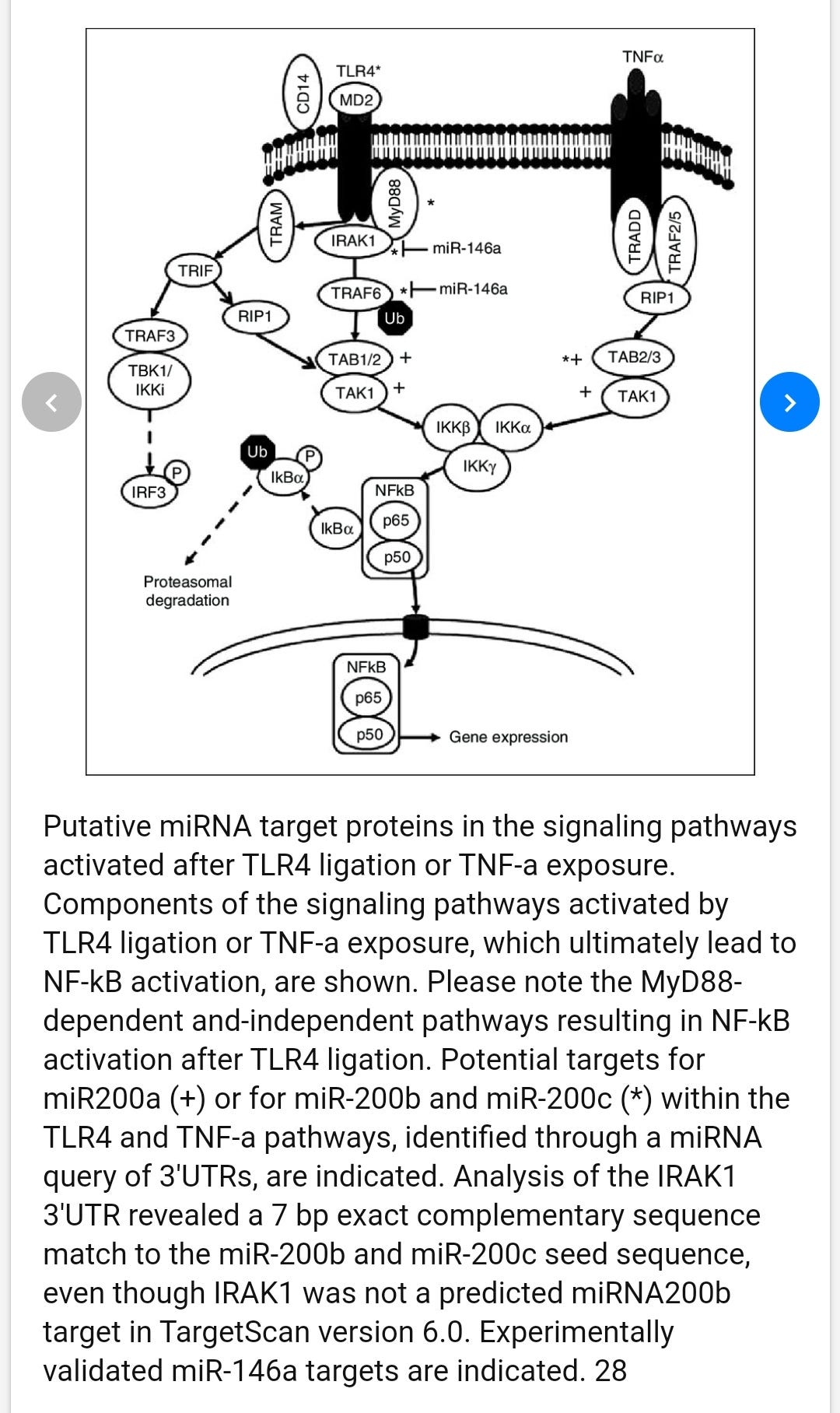
The role of MicroRNAs miR-200b and miR-200c in TLR4 signaling and NF-kappa B activation
Abstract and Figures
Recognition of microbial products by members of the Toll-like receptor (TLR) family initiates intracellular signaling cascades that result in NF-κB activation and subsequent production of inflammatory cytokines. We explored the potential roles of microRNAs (miRNAs) in regulating TLR pathways. A target analysis approach to the TLR4 pathway adaptor molecules identified several putative targets of miR-200a, miR-200b and miR-200c. miRNA mimics were co-transfected with a NF-κB activity reporter plasmid into HEK293 cells stably expressing TLR4 (HEK293-TLR4). Mimics of both miR-200b and miR-200c, but not miR-200a, decreased NF-κB reporter activity in either untreated cells or in cells treated with endotoxin:MD2 as a TLR4 agonist. Transfection of HEK293-TLR4 cells with miR-200b or miR-200c significantly decreased expression of MyD88, whereas TLR4, IRAK-1 and TRAF-6 mRNAs were unaffected. When miR-200b or miR-200c mimics were transfected into the differentiated monocytic THP-1 cell line, the abundance of MyD88 transcripts, as well as LPS-induced expression of the pro-inflammatory molecules IL-6, CXCL9 and TNF-α were diminished. These data define miRNAs miR-200b and miR-200c as factors that modify the efficiency of TLR4 signaling through the MyD88-dependent pathway and can thus affect host innate defenses against microbial pathogens.
MicroRNA Roles in the Nuclear Factor Kappa B Signaling Pathway in Cancer (2018)
Abstract
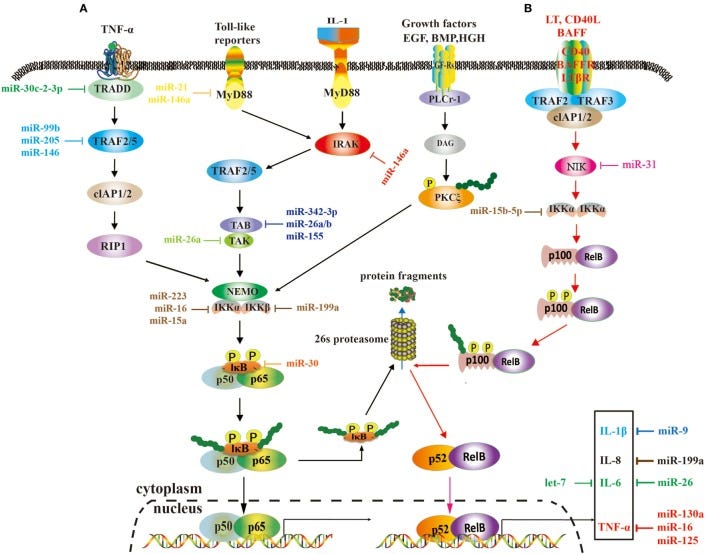
Nuclear factor kappa B (NF-κB) is a pluripotent and crucial dimer transcription factor that orchestrates various physiological and pathological processes, especially cell proliferation, inflammation, and cancer development and progression. NF-κB expression is transient and tightly regulated in normal cells, but it is activated in cancer cells. Recently, numerous studies have demonstrated microRNAs (miRNAs) play a vital role in the NF-κB signaling pathway and NF-κB-associated immune responses, radioresistance and drug resistance of cancer, some acting as inhibitors and the others as activators. Although it is still in infancy, targeting NF-κB or the NF-κB signaling pathway by miRNAs is becoming a promising strategy of cancer treatment.
A role of microRNAs (miRNAs) in the nuclear factor kappa B (NF-κB) signaling pathways. These two pathways rely on tumor necrosis factor (TNF), toll-like, interleukin (IL)-1, and EGF receptors (A), and BAFF reporter and CD40 (B), respectively, and they are activated or repressed by miRNAs. Activated NF-κB promotes or restrains the expression of tumor-associated genes.
MicroRNAs (miRNAs) are a set of non-coding small RNA molecules in control of gene expression at posttranscriptional/translational level. They not only play crucial roles in normal developmental progress, but also are commonly dysregulated in human diseases, including cancer. MiR-200 is a family of tumor suppressor miRNAs consisting of five members, which are significantly involved in inhibition of epithelial-to-mesenchymal transition (EMT), repression of cancer stem cells (CSCs) self-renewal and differentiation, modulation of cell division and apoptosis, and reversal of chemoresistance. In this article, we summarize the latest findings with regard to the tumor suppressor signatures of miR-200 and the regulatory mechanisms of miR-200 expression. The collected evidence supports that miR-200 is becoming a new star miRNA in study of human cancer.
Recently, Zhu et al. reported that the miR-200bc/429 cluster was impaired in vincristine-resistant gastric cancer cells and cisplatin-resistant lung cancer cells. However, the restoration of the miR-200bc/429 cluster could sensitize the tumors cells to chemotherapy. The mechanistic studies suggested that this effect resulted from, at least partially, recurred apoptosis led by the suppressive effect of miR-200 on two anti-apoptotic factors, B-cell lymphoma 2 gene (BCL-2) and X-linked inhibitor of apoptosis protein gene (XIAP)
2. Tumor suppressive signatures of miR-200
2.1. Inhibition of epithelial mesenchymal transition (EMT) and tumor metastasis
The involvement of miR-200 in cancer originates from several studies demonstrating a significant role for miR-200 in EMT, which is an important process in tumor progression as well as embryonic development. The expression of miR-200 family members was found to be highly associated with the epithelial phenotype of cancer cells, serving as a class of key markers for E-cadherin-positive and vimentin-negative cancer cell lines [25-27]. ZEB1 and ZEB2 are two members of the zinc-finger E-box binding homeobox family, and they have been defined as the master regulators in EMT [24]. The mechanism by which ZEB1 and ZEB2 facilitate EMT is that ZEBs can efficiently inhibit the cell-cell adhesion molecule E-cadherin, given that the aberrant expression of E-cadherin is a hallmark of EMT [28]. A number of studies have documented that, in epithelial cancer cells, highly expressed miR-200 represses the expression of ZEBs; whereas in mesenchymal cancer cells, impaired expression of miR-200 leads to induction of ZEBs and subsequent repression of E-cadherin [25-27]. Following the studies on the ability of miR-200 to regulate the expression of ZEB1 and ZEB2, dozens of others were carried out in various types of human tumors as summarized in Table 1[29-60]. While expression of the miR-200 family members was determined to be impaired in various human tumor cells leading to EMT and disease progression, increased miR-200 family member expression led to a reversal of EMT in bladder cancer, gastric cancer, nasopharyngeal carcinomas, ovarian cancer, pancreatic cancer, and prostate cancer [29, 39, 40, 44, 46, 52, 54]. In support of these results, studies by using clinical patient samples also indicated strong correlations between miR-200 expression and tumor progression in a variety of tumor types[31-33, 35, 37, 39, 41, 42, 45, 47, 48, 50, 53, 54, 56, 58-60]. Furthermore, it has been noted that low level expression of miR-200 could correlate with poor survival and serve as a prognostic marker for cancer patients [29, 38, 47, 58-60]. As such, the findings from the studies of miR-200 and EMT have become the benchmarks for further research on the tumor suppressive signatures of miR-200.
Figure 1
Tumor suppressive signatures of miR-200. (A) MiR-200 inhibits EMT by interacting with ZEB1/2 and the Notch pathway. (B) MiR-200 represses self-renewal and differentiation in CSCs. (C) MiR-200 is involved in the regulation of cell division and apoptosis.
Dysregulation of miR-200 in various human cancers
EMT is considered the initiating event for cancer metastasis. Two different groups simultaneously reported that miR-200 was significantly downregulated in metastases and metastatic-like primary tumors, thereby relieving the repression conferred by the mesenchymal transcription factor ZEB1 in vivo. Forced expression of miR-200 abrogated the capacity of tumor cells to undergo invasion and metastasis, underscoring the role for miR-200 in the regulation of both EMT and subsequent metastases [61, 62]. A similar conclusion was drawn through studying different human ovarian cancer cell lines with distinct capabilities to metastasize [63], while miR-200 has also been implicated in the reversal of the metastatic phenotype of non-small cell lung cancer, as its re-expression has been shown to downregulate the expression of many prognostic markers for metastasis, such as alpha thalassemia/mental retardation syndrome X-linked gene (ATRX), deleted in liver cancer 1 gene (DLC1), hereditary hemochromatosis gene (HFE), and heterogeneous nuclear ribonucleoprotein A3 gene (HNRNPA3)[43]. Given the mechanistic studies supporting that the Notch signaling pathway plays a crucial role in the regulation of EMT and thus metastasis during cancer progression [64], miR-200 was found to decrease expansion of human metastatic prostate cancer cells by targeting the Notch ligand Jagged1 and the mastermind-like coactivators Maml2 and Maml3, the key components in the Notch pathway [65, 66]. Complementary to these results, Yang et al. found that the Notch ligand Jagged2 was also able to inhibit miR-200 family expression at the transcriptional level by induction of GATA transcription factors, which eventually led to promotion of tumor metastasis in vivo [67]. These findings support a regulatory loop consisting of miR-200 and the Notch signaling pathway; the balance of their interaction can potentially decide the stages of tumor progression (Figure 1A). However, the molecular mechanisms accounting for the tumor suppressor roles of miR-200 are still largely unknown, although the Notch signaling pathway sheds light on understanding of its anti-metastatic activity. A recent study reported the controversial evidence that miR-200 was found to be upregulated in breast cancer 4T1 cells that formed macroscopic metastases in vivo when compared with related cells invading distant tissues but were unable to colonize. The authors proposed that miR-200 might be involved in promotion of the last step of the metastatic cascade when establishing macroscopic metastatic masses at distant sites [68]. Given most current findings supporting miR-200 as a tumor suppressor, additional evidence is needed to confirm such a hypothesis in which miR-200 plays an oncogenic role. Together with the myriad of aforementioned studies from a wide variety of cancers listed in Table 1, these data suggest that the miR-200 family plays a significant role in combating not only EMT, but also tumor cell invasion and metastases. Thereby, miR-200 has great potential to become a novel class of biomarkers for tumor prognosis and targets for new drug development against tumor progression.
We need to be particularly aware of methylation as longer term deactivation of the tumor suppressors could result, just as we see from long term smoking:
Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells
Abstract
Background: The miR-200c/141 cluster has recently been implicated in the epithelial to mesenchymal transition (EMT) process. The expression of these two miRNAs is inversely correlated with tumorigenicity and invasiveness in several human cancers. The role of these miRNAs in cancer progression is based in part on their capacity to target the EMT activators ZEB1 and ZEB2, two transcription factors, which in turn repress expression of E-cadherin. Little is known about the regulation of the mir200c/141 cluster, whose targeting has been proposed as a promising new therapy for the most aggressive tumors.
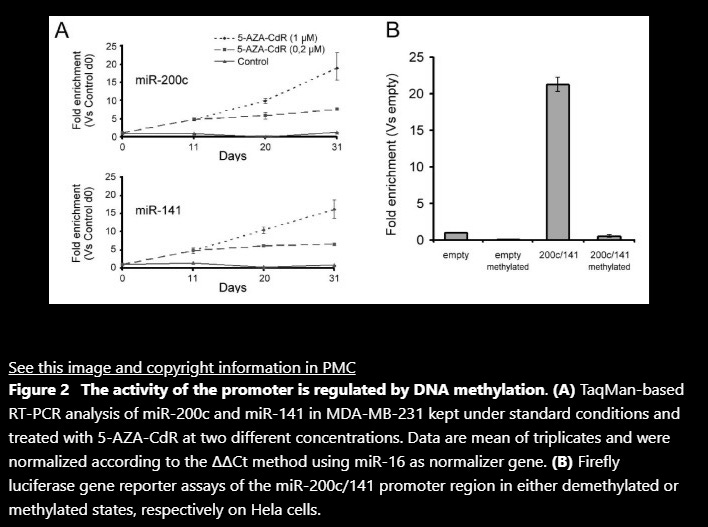
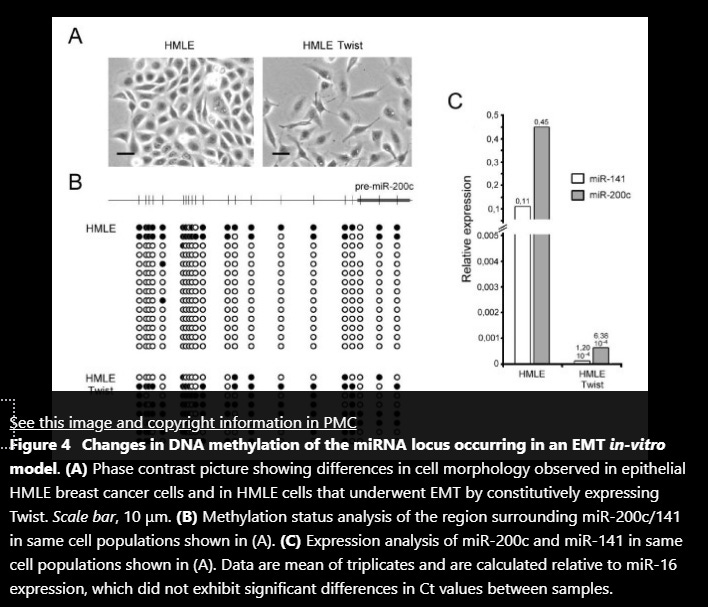
Findings: We show that the miR-200c/141 cluster is repressed by DNA methylation of a CpG island located in the promoter region of these miRNAs. Whereas in vitro methylation of the miR-200c/141 promoter led to shutdown of promoter activity, treatment with a demethylating agent caused transcriptional reactivation in breast cancer cells formerly lacking expression of miR-200c and miR-141. More importantly, we observed that DNA methylation of the identified miR-200c/141 promoter was tightly correlated with phenotype and the invasive capacity in a panel of 8 human breast cancer cell lines. In line with this, in vitro induction of EMT by ectopic expression of the EMT transcription factor Twist in human immortalized mammary epithelial cells (HMLE) was accompanied by increased DNA methylation and concomitant repression of the miR-200c/141 locus.
Conclusions: The present study demonstrates that expression of the miR-200c/141 cluster is regulated by DNA methylation, suggesting epigenetic regulation of this miRNA locus in aggressive breast cancer cell lines as well as untransformed mammary epithelial cells. This epigenetic silencing mechanism might represent a novel component of the regulatory circuit for the maintenance of EMT programs in cancer and normal cells.
More on B-cell lymphoma 2 gene (BCL-2), which promotes anti-apoptosis and is suppressed by MiR-200 (until it isn't). Over to Wikipedia:
B-cell lymphoma 2 gene (BCL-2)
Role in disease
Damage to the Bcl-2 gene has been identified as a cause of a number of cancers, including melanoma, breast, prostate, chronic lymphocytic leukemia, and lung cancer, and a possible cause of schizophrenia and autoimmunity. It is also a cause of resistance to cancer treatments.[13]
Cancer
Cancer can be seen as a disturbance in the homeostatic balance between cell growth and cell death. Over-expression of anti-apoptotic genes, and under-expression of pro-apoptotic genes, can result in the lack of cell death that is characteristic of cancer. An example can be seen in lymphomas. The over-expression of the anti-apoptotic Bcl-2 protein in lymphocytes alone does not cause cancer. But simultaneous over-expression of Bcl-2 and the proto-oncogene myc may produce aggressive B-cell malignancies including lymphoma.[14] In follicular lymphoma, a chromosomal translocation commonly occurs between the fourteenth and the eighteenth chromosomes – t(14;18) – which places the Bcl-2 gene from chromosome 18 next to the immunoglobulin heavy chain locus on chromosome 14. This fusion gene is deregulated, leading to the transcription of excessively high levels of Bcl-2.[15] This decreases the propensity of these cells for apoptosis. Bcl-2 expression is frequent in small cell lung cancer, accounting for 76% cases in one study.[16]
Auto-immune diseases
Apoptosis plays an active role in regulating the immune system. When it is functional, it can cause immune unresponsiveness to self-antigens via both central and peripheral tolerance. In the case of defective apoptosis, it may contribute to etiological aspects of autoimmune diseases.[17] The autoimmune disease type 1 diabetes can be caused by defective apoptosis, which leads to aberrant T cell AICD and defective peripheral tolerance. Due to the fact that dendritic cells are the immune system's most important antigen-presenting cells, their activity must be tightly regulated by mechanisms such as apoptosis. Researchers have found that mice containing dendritic cells that are Bim -/-, thus unable to induce effective apoptosis, suffer autoimmune diseases more so than those that have normal dendritic cells.[17] Other studies have shown that dendritic cell lifespan may be partly controlled by a timer dependent on anti-apoptotic Bcl-2.[17]
Other
Apoptosis plays an important role in regulating a variety of diseases. For example, schizophrenia is a psychiatric disorder in which an abnormal ratio of pro- and anti-apoptotic factors may contribute towards pathogenesis.[18] Some evidence suggests that this may result from abnormal expression of Bcl-2 and increased expression of caspase-3.[18]






Bravo!!