


Walkthrough: "mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues" (22rd June '23)
And why there is no such thing as an anti-cancer "vaccine"

Reading time: about 60 minutes if you take it slow, according to Substack stats.
Also available in other languages:
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.

Introduction
I will continue to work on narrative literature reviews which, by their very nature are difficult to foreshorten without leaving out too many important papers but in recognition of the time commitment required to follow my work I will be returning to more frequent but shorter posts which marked the origins of this Substack, at least that’s my goal!
Future Substacks will consider the often overlooked but critically important mineral magnesium and pathologies associated with the “scavenger protein” CD163.
I will also venture into the physiology of hydration and a longer multi-part review of a very useful therapeutic icariin and its metabolites, - watch this space.
This Substack will be a walkthrough of a newly published, peer reviewed paper by Dr. Helene Banoun from the journal site MDPI and appearing in their special Issue "The Future of Drug Discovery and Development".
About the author, Dr Banoun is an independent researcher, worked for the French Institute of Health and Medical Research from 1982-86 and is cited in 87 publications, according to ResearchGate.
https://www.researchgate.net/profile/Helene-Banoun
You can follow Helene here:
https://twitter.com/BanounHelene
This paper is a narrative review, is quite candid in its observations and citations, and definitely worth highlighting for subscribers. Its quite surprising and reassuring that it managed to get past the “retraction” censors. This is becoming harder and harder to justify as the research papers, VAERS reports and case studies continue to mount. Its also a warning to pause the ongoing approach of using fast-tracked 100 day experimental mRNA tech as a snake oil to treat many diseases and illnesses and cancers, as well as reformulating traditional “vaccines” in this synthetic form.
G7 Leaders welcomed the 100 Days Mission (100DM) at the June Carbis Bay Summit, and committed to working together, between sectors and across national borders, to deliver this ambitious Apollo mission. Leaders spoke of the unpredictability of future health emergencies and emphasised the need to harness scientific innovation and public-private collaboration to develop an armamentarium of diagnostics, therapeutics and vaccines (DTVs) available within the first 100 days of a future pandemic threat being detected, consistent with our core principles around equitable access and high regulatory standards.
Extract from “1OO DAYS MISSION” (2nd December 2021)
And in the UK from legislative changes announced in March ‘23, we can see that the intention is for long term clinical trials to be a thing of the past. Buyer beware!
MHRA’s Chief Executive Dr June Raine added: “introduction of the new routes will complement the work being done through the MHRA’s Innovative Licensing and Access Pathway (ILAP), establishing an additional avenue for accelerated access to life-saving new medicines.”
Through ILAP, the MHRA Innovation Passport was awarded to Boehringer Ingelheim’s investigational BI 907828 in November 2022, for de-differentiated liposarcoma (DDLPS). This is a rare tumoural cancer which has not seen new first-line treatment in over forty years. Read the article on Boehringer Ingelheim’s ILAP designation here.
In March 2023, the HM Treasury announced a total of £10 million was awarded to the MHRA, to support development of the new recognition framework. This will help bring innovative new medicines such as cancer vaccines and AI-based therapeutics for mental ill-health to UK patients more quickly
New recognition routes to fast-track UK medicine access
As my particular ongoing interest is within the field of carcinogenesis I will focus particularly on these aspects but encourage you to read the whole paper in full at your leisure.
On this point those sigma numbers are indicative of a flock of black swans:
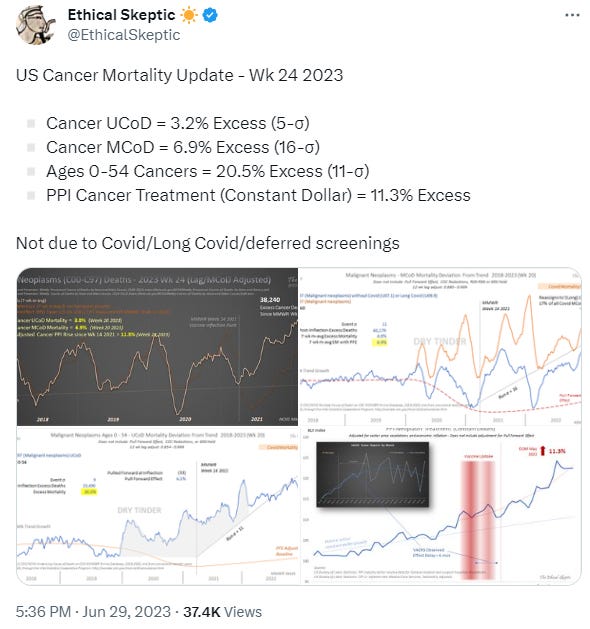
As ever I will post the abstract and conclusions in full and add interpretations and additional content where appropriate.
mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues1
Abstract
COVID-19 vaccines were developed and approved rapidly in response to the urgency created by the pandemic. No specific regulations existed at the time they were marketed. The regulatory agencies therefore adapted them as a matter of urgency. Now that the pandemic emergency has passed, it is time to consider the safety issues associated with this rapid approval. The mode of action of COVID-19 mRNA vaccines should classify them as gene therapy products (GTPs), but they have been excluded by regulatory agencies. Some of the tests they have undergone as vaccines have produced non-compliant results in terms of purity, quality and batch homogeneity. The wide and persistent biodistribution of mRNAs and their protein products, incompletely studied due to their classification as vaccines, raises safety issues. Post-marketing studies have shown that mRNA passes into breast milk and could have adverse effects on breast-fed babies. Long-term expression, integration into the genome, transmission to the germline, passage into sperm, embryo/fetal and perinatal toxicity, genotoxicity and tumorigenicity should be studied in light of the adverse events reported in pharmacovigilance databases. The potential horizontal transmission (i.e., shedding) should also have been assessed. In-depth vaccinovigilance should be carried out. We would expect these controls to be required for future mRNA vaccines developed outside the context of a pandemic.
Keywords:
mRNA vaccines; COVID-19; drug regulation; gene therapy; vaccinovigilance; pharmacokinetics
From the introduction. You are the clinical trial as GTP regulations do not apply. Its a bit like needing to pass legislation in order to read it:
The anti-COVID-19 mRNA vaccines are the first mRNA vaccines marketed. mRNA vaccines, which represent a new class of vaccine, should be subject to more controls than conventional vaccines because they are based on several new technologies [1]. Although incompletely defined, the mode of action of mRNA vaccines [2] should classify them as gene therapy products (GTP) [3]. But mRNAs as vaccines against an infectious disease have been excluded from GTP regulation by US and EU regulations [4]. No specific regulations existed before the year 2020 for mRNA vaccines. “The current guidelines either do not apply, do not mention RNA therapeutics, or do not have widely accepted definition” [5]. Regulatory agencies therefore had to adopt an emergency procedure to monitor the testing of these products, the rolling review. In rolling reviews, data are submitted and reviewed as they become available before the full data package is available and specific controls for this new platform have been requested [6].
Fast tracked cancer vaccines with approvals based on very limited studies. These work, in theory, by addressing to specific areas2:
Presenting tumour antigens to the immune system.
Stimulating anti-tumour immunity.


Shortcomings to this approach may include failure due to:
Extrinsic and intrinsic resistance to mechanisms of immunity, including tumour cell genetic mutations.
Exhaustion, anergy or full immunosenescence of the patient,
Risk of autoimmune attack against healthy tissues due to overlapping epitopes.
Failure to address many other cancer signalling pathways, including from viruses or systemic inflammation.
Antigenic variability.
Original antigenic sin in the vaccine primed response to the tumour.
Data from infectious disease research has demonstrated that even minimal differences or overlap in amino acids between mutated and corresponding wild-type epitopes dramatically narrow the emerging TCR repertoire, rendering T cells dysfunctional, unresponsive and/or exhausted (Klenerman and Zinkernagel 1998). Original antigenic sin (OAS) is a well-documented phenomenon, initially described in influenza as imprinting (memory induction) by the initial viral infection on the antibody response to subsequent vaccination or infection with a mutant virus (Monto et al. 2017; Vatti et al. 2017). Despite long debates, there is now convincing evidence that reduced vaccine effectiveness (estimated as 40–60%) is related to OAS (Monto et al. 2017). This phenomenon was later described for cytotoxic T lymphocyte (CTL) responses, where mice primed with lymphocytic choriomeningitis virus WE strain (LCMV-WE) were not able to generate an adequate CTL response against a mutated LCMV-WE virus, bearing a variant CTL epitope during a subsequent infection. These effects led to immune escape and impaired clearance of mutant viruses (Klenerman and Zinkernagel 1998). One of the most alarming cases was caused by the Sanofi Pasteur Dengue vaccine, which sensitized some of the dengue-naïve recipients to severe life-threatening disease. This effect was caused by antibody-dependent enhancement, an OAS-related phenomenon (Shukla et al. 2020). The possible involvement of OAS, a strictly pathogen-related phenomenon, unappreciated in cancer immunology, might contribute to the final outcome of tumor-immune interactions. To elucidate our hypothesis that OAS may have a detrimental role in cancer evolution and immune escape (Domínguez-Romero et al. 2020), we have generated preliminary experimental data (currently still in process), which highly suggests the presence of this phenomenon in cancer.
…Despite the acknowledgment, for decades now, of the continuous mutations in tumor cells and the acceptance of cancer as a genetic evolutionary disease, current vaccine concepts are not addressing these critical issues adequately. At this point, only out-of-box thinking can redirect scientific innovation away from the current paradigm and may propel it towards authentic and viable alternatives.
NeoAg: Neoantigen single-use cancer vaccine candidates.
TAAs: Tumour-associated antigens
Few cancer types have been targeted thus far for NeoAg-based treatment; though, we could expect similar results from any one of the more than 200 cancer types if the indications for this immunotherapy modality are expanded. However, NeoAg-based vaccines have already made one significant “contribution” in the field of cancer research and therapy; they have dramatically reduced the number of clinical trials using TAAs, oncogenes and other non-mutated antigens. This will free up human and financial resources and redirect them towards the development of alternative vaccine strategies. Kissick stated years earlier that if NeoAgs are not capable of inducing/expanding more significant numbers and more diverse repertoires of T cells with increased specificity for tumor Ags, than those obtained upon vaccination with TAAs, “it will certainly not be worth the significant extra effort required to generate these individualized vaccines” (Kissick 2018): we firmly believe that the validity of this opinion still holds true.
Manoutcharian K, Guzman Valle J, Gevorkian G. Neoantigen Cancer Vaccines: Real Opportunity or Another Illusion? Arch Immunol Ther Exp (Warsz). 2021;69(1):12. doi:10.1007/s00005-021-00615-8/www.ncbi.nlm.nih.gov/pmc/articles/PMC8080209/
Early trials also showed they weren’t suited to treating advanced cancers, yet this is the area where effective treatments are really needed. I list a few here and the cancer pathways targeted, if you haven’t seen them already:
With regulatory hurdles out the way there is something of an mRNA “vaccine” pharma gold rush in progress right now.

Potential safety issues arising from the absence of these controls will be discussed. This is all the more urgent as manufacturers are planning to replace certain “classic” vaccines with mRNA vaccines [2], starting with influenza vaccines. Indeed, Sanofi is launching a clinical trial of the first mRNA-based seasonal flu vaccine candidate [7] and Moderna has many mRNA vaccines in clinical trials (COVID-19, influenza, human metapneumovirus, parainfluenzas, RSV, HCoV, CMV, EBV, HSV, varicella, herpes, HIV, Zika, Nipah), in particular a phase 3 trial of the flu vaccine [8].
A phase 1 clinical trial is being launched for an mRNA-LNP influenza vaccine [9]. For these flu vaccines, emergency approval should not apply and the requirement for these additional studies should not be exceeded.
In addition, cancer “vaccines” are being announced (e.g., Moderna and Merck are partnering in trials of mRNA-4157/V940, an anti-melanoma “vaccine” combined with Keytruda—a monoclonal antibody directed against the programmed cell death receptor, PD-1) that acts by enhancing the ability of the body’s immune system to detect and fight tumor cells, by blocking the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating the anti-T cell response, particularly the antitumor response [10]).
We must be very vigilant about the term vaccine associated with therapeutic drugs, particularly with regard to the regulations that apply to them. These therapeutics are not vaccines against infectious diseases and must therefore continue to comply with GTP regulations.
The FDA define them as gene therapies, the one definition they cannot meet is that of a vaccine. A new description should be that they are a kind of pro-drug where the active ingredient is synthesised in vivo and stricter regulations should therefore apply:
According to the FDA [12], gene therapy is a medical intervention based on the modification of the genetic material of living cells. Cells may be altered in vivo by gene therapy given directly to the subject.
According to European regulations, vaccines are products capable of producing active immunity [17] and contain antigens capable of inducing active immunity against an infectious agent [4]. According to the EMA [11], the active substance of the COVID-19 Pfizer vaccine is mRNA: it is not an antigen. Therefore, according to the European and French pharmacopoeias, mRNAs should not be considered as vaccines because they do not contain antigens.
Moreover, still according to their mode of action, mRNA vaccines can be considered as pro-vaccines; this is a neologism modelled on the word pro-drug which designates a drug which, after administration, is converted by the organism into a pharmacologically active drug. In fact, according to the principle of mRNA, this must be translated into protein by the cells of the person vaccinated (the injected substance is not the substance causing an active immunization). According to the FDA [18], mRNA vaccines correspond to the TypeIA of pro-drugs, which are substances that are converted by cells into active drugs. This pro-drug property could imply additional controls to those applied to vaccines. However, neither the FDA nor the EMA make any reference to these qualifications for mRNA anti-COVID-19 vaccines.

With regard to contamination with SV40 promoter containing plasmids, as discovered by McKernan, K et al and others in 20233:
In a 1996 document [21] on DNA plasmids for preventive infectious disease indications, it is specified (as for all FDA guidelines) that no subsequent requirements were established: these were non-binding recommendations. For this reason, this study will be based primarily on EMA documents; although EMA guidelines are not legally binding, applicants need to provide justification for any deviations.
There can be no such thing as an “anti-cancer vaccine” if you aren’t targeting an infectious disease. As an aside I wish they would target latent onco-viruses such as polyomaviruses too (see my last Substack from 9th June), but optimal outcomes aren’t their concern it would appear:
The upshot being that these products managed to fall between strict regulatory regimes for either gene therapy products or vaccines:
2.4. Why Are mRNA Vaccines Excluded from the Regulation of Gene Products?
According to Guerriaud and Kohli [4], “it is difficult to answer with certainty why vaccines against infectious diseases have been excluded. The definition [of vaccines] has not changed since 1975, a period when there was no “vaccine” against cancer” [17]; they are agents capable of producing active immunity against an infectious disease. At that time, the only existing vaccines were against infectious diseases and the current definition of a vaccine is limited to an immunological drug against an infectious disease. Therefore, an anti-cancer drug can, in no way, be called a “vaccine”. It should be noted that therapeutic AIDS vaccines, based on lentiviruses and acting as gene therapies, because they integrate into the genome, have also been excluded from gene therapies [29]. It can be assumed that the applicant has argued that the product has both a therapeutic and a prophylactic mode of action, but the document is not available on the EMA website.
From a public health point of view, and knowing that anti-COVID-19 mRNAs considered as vaccines have not undergone all the strict controls required for GTPs (see below), one could object that a product intended for the majority of the world’s healthy population should be subject to more stringent regulation than a GTP intended for a few rare people suffering from a rare disease or cancer (this time concerning millions of people). Moreover, according to the EMA [28], “Since vaccines in most cases are given to large numbers of healthy individuals, there is a need for a solid nonclinical safety evaluation”.
Although they are are de facto pro-drugs, the regulators class them as mRNA vaccines, so they get a free pass:
2.6. Controls Required as a Pro-Drug
The FDA [18] points out the particular problems of pro-drugs raised by their more or less complete conversion into an active substance and the question of toxicity. According to the FDA, it is necessary to define how the pro-drug contributes significantly to the toxicity profile of the active drug, particularly as a function of the site of transformation and action. For mRNA vaccines, biological transformation occurs in many cell types and in all organs (see below), whereas the desired goal, i.e., immunization, will only occur in immune cells. However, mRNA vaccines are not classified as pro-drugs and therefore do not have to undergo the controls concerning the site of transformation and action.
The manufacturers failed to provide the requested product quality control safety data to the EMA within the required timescales (never, as it turned out), but their licence wasn’t pulled with the same vigour that unapproved researched papers get retracted.
I went through this car crash of regulatory capture back in January ‘22:
Despite the absence of specific regulations for mRNA vaccines, the EMA has added the following controls on product quality: identity (by RT-Sanger sequencing for Spikevax and Next Generation sequencing for Comirnaty); total RNA content (UV), purity (RP-HPLC); product-related impurities (RP-HPLC); % 5′ Capped (RP-UPLC); % PolyA tailed RNA (RP-HPLC for Spikevax, not fully described for Comirnaty) and residual DNA template (qPCR).
However, some specific obligations reported in the first report (for example, the mode of action, which is not described) [11,33,34] have not been fulfilled according to the 2021 report [35]. Regarding impurities, the method of determination of bacterial endotoxin should be specified [11,33,34]. The levels of endotoxin found are not specified in the documents of the Australian regulatory authority, the TGA, and some batches were still under evaluation at the time of batch release [36]. The accepted limit is 12.5 EU/mL [34].
Concerning the quality of the product, the in vitro transcription method is not sufficiently described and the characterization of the mRNA is not satisfactory; the methods of evaluation of truncated and modified mRNAs must be more detailed. A potency test is not satisfactory, in vitro expression test needs to be updated. If the Poly(A) tail length and percentage partly remains, the REC20 is not fulfilled. If 3 months of stability data are provided, 6 months of data are expected [36], and the presence of truncated mRNA and truncated protein expression is not sufficiently explored [33]. EMA calls for a clarification of the mode of action [11,33,34].
Therefore, the results of the controls required for GTPs that were requested by the EMA are not sufficient according to the EMA reports.
Advanced in vivo or human pharmacokinetics studies were not conducted, despite going against EMA and CBER guidance, as well as the precautionary principle:
Concerning the US-FDA, one should refer to the CBER (Center for Biologics Evaluation and Research) guide in charge of regulating these products, which only issues non-binding recommendations [39], as well as to the 2013 instructions [20], which, globally, impose the same criteria as the EMA.
3.2. Pharmacokinetics
According to the EU, GTMPs require specific tests or trials to evaluate the risk of genome integration and germ-line transmission [24,38], even if this integration is unlikely [37], as it is the case for RNA.
GTMPs require specific tests or trials to evaluate the risk of insertional mutagenesis, tumorigenicity, embryo/fetal and perinatal toxicity and long term expression [38] that have not been performed.
Why were the products not suspended pending further safety studies when their failure at the approved 2 doses became apparent?
For GTMPs, the EMA requires extensive studies on both the nucleic acid and the vector particle/delivery system that includes biodistribution, dose study, potential target toxicity, the identification of the target organ to obtain biological activity, toxicity linked to the expression of structurally altered proteins, reproductive toxicity (for these studies, tests must follow the ICH M3 document [40]), repeated toxicity and excretion in the environment. These studies are required for products containing DNA because the document was drafted in 2006 [37] and mRNA vaccines were not considered at the time. Repeated toxicity was not adequately studied because only two doses of vaccine were planned [32].
It is necessary to insist on pharmacokinetic studies, which are generally not required for vaccines unless they are based on a new formulation or when the vaccine contains novel adjuvants or excipients. The need for such studies must be assessed for vaccines on a case-by-case basis by the regulatory authorities [23]. Moreover, “the standard absorption/distribution/metabolism and excretion studies for conventional medicinal products may not be relevant for GTMPs” [38]. For example, the route of administration that is considered as the worst case scenario (e.g., intravenous, representing the effect of widespread dissemination of the GTMP) should be considered. For GTPs, shedding studies are expected that address excretion and dissemination in the body, including studies on persistence, clearance and mobilization. Biodistribution studies should also address pharmacokinetic studies of the transgenic product (e.g., expressed proteins). The studies provided by the manufacturers seem to be incomplete from this point of view (see Section 4).
Carcinogenic potential just wasn’t considered, although from another paper I am contributing to (hat tip to lead author Genervter @NarfGB, @LadyBug, @DrBine, Jo, Annelise and Typhaine) the parties involved must have known that canonical MAPK and other cancer pathways were being signalled to a significant degree, and for extended time periods:
3.3. Controls on Biological Drugs Not Carried Out
The EMA, like the European Commission, considers that “RNA-derived products should be considered as biological products, even if they are not derived from a biological source” [41]. According to European regulations [17], for a biological drug, a list of biological activities must be provided, and studies of reproductive function, embryo-fetal and perinatal toxicity and mutagenic and carcinogenic potential must be considered. We have seen above that these tests have not been carried out, and that the biological activities of the active ingredient—mRNA—have not been sufficiently described.
Genotoxicity issues, including insertional mutagenesis and consequent tumorigenicity, should be evaluated carefully in relevant in vitro/in vivo models. Immune suppression, a causative factor for tumorigenesis in humans, must be investigated. According to Spikevax-EPAR, no carcinogenicity, insertional mutagenensis nor tumorigenicity in in vivo studies were submitted. Embryo-foetal and perinatal toxicity studies may be required if women of child-bearing potential are to be exposed to GTMPs [38].
Most cancer latencies are in the 10-30 year range, so surveillance for shorter durations than this is only likely to detect cases of recurrence or low latency soft tissue cancers such as lymphomas, and most of these will probably go unrecorded as unrelated to the GTP’s that the patient received. Within 42 days all but the most aggressive turbo-cancer responses would go unrecorded:
Studies have suggested that the latency period for ovarian cancer is between 30 and 40 years [13], [14], [15]. For colorectal cancer, it is thought to take 5–10 years from initiation to adenoma development and 5–15 years from adenoma to invasive disease [16].
Nadler and Zurbenko applied a Weibull survival model to estimate the approximate length of time between biological initiation to cancer diagnosis for 44 cancer types [14]. Overall, over 35 of these cancer types were estimated to develop at least 10 years before cancer diagnosis, ranging from 6.6 to 57 years for solid tumors and 2.2 and 35.7 years for lymphoproliferative cancers, highlighting the wide variability across cancer types.
Indeed, while most cancers typically have long latency periods, there are some examples of more rapid cancer development following exposure, particularly for hematopoietic cancers, which appear to have much shorter induction periods [4], [35].
…The evaluation of drug–cancer associations, particularly those with new medications that emerge from RCTs or case reports with short exposure periods, is complicated by the fact that often the mechanisms underlying cancer associations are unknown. Researchers may be too quick to dismiss associations observed within shorter periods of time as noncausal [59]. Indeed researchers should be mindful of falsely declaring a medication safe. Walker suggests that “the observation is the hypothesis” and argues the appropriate response is to test the signal in a similar setting under controlled circumstances with sufficient statistical power [59]. However, it is difficult to draw conclusions in the absence of a biological model, as is often the case. Additionally, hypotheses of carcinogenesis also commonly arise from pharmacology and studies after drug approval. In general, while the information that generated the signal is important, so too is our understanding of cancer biology and latency to help inform assumptions. Overall, examples of tumor promotion effects are limited, and given the typically long latency of cancer, most cancer–drug associations are considered to have longer latency periods4.
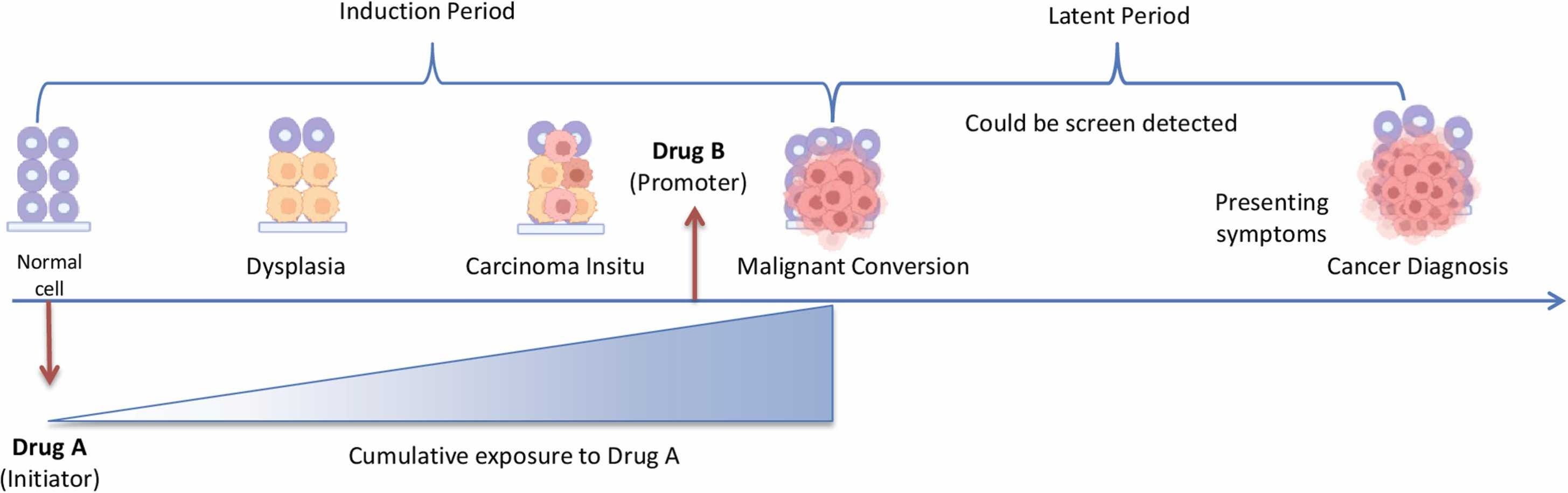
3.5. Vaccinovigilance
According to the EMA, GTMPs have an obligation to provide safety and efficacy data for 30 years after the expiry date of the drug, which is beyond the requirements of classical pharmacovigilance [24].
According to the FDA regulations for GTPs [42], a long-term follow-up of adverse events must be performed for at least 5 years for new clinical conditions, such as: new malignancy(ies), new incidence or exacerbation of a pre-existing neurologic disorders, new incidence or exacerbation of a prior rheumatologic or other autoimmune disorder, new incidence of a hematologic disorder and new incidence of infection (potentially product-related).
According to the European regulation [13], a strategy for the long-term follow-up of safety and efficacy shall be included in the risk management plan.
With regard to conventional vaccines, the duration of observation of adverse events is generally only a few weeks. The Brighton Collaboration [43], which is responsible for monitoring the safety profiles and benefit/risk ratios of vaccines, has published a guide for monitoring the selected adverse events of vaccines in general. Follow-up times are sometimes specified but rarely exceed 2 months [44] and, according to an FDA COVID-19 vaccine pharmacovigilance study [45], vaccinated individuals are followed for up to 42 days.
4. Discussion
4.1. Controls Required for a Pro-Drug That Have Not Been Carried Out
If anti-COVID-19 mRNAs had been classified as pro-drugs, they would have had to undergo controls concerning the site of transformation and action [18]. It would thus have been detected that the spike protein translated from the mRNA is not only found in the immune cells of the muscle where the mRNA is injected.
Your box of cornflakes is manufactured to much higher quality control standards than these mass-injected gene therapy products. Even then you risk acrylamide, mycotoxin and pesticide contamination if you buy the wrong brand, but that’s the food industry for you5.

Without a proteomic analysis of the expressed proteins, truncated or otherwise, it is challenging to predict the potential pathologies that may result, but we can shortlist some:
Failure to mount sufficient antiviral immune response (for better or worse).
Risk of initiating autoimmune disorders, as detailed here:
Risk of protein misfolding. These are linked to a variety of disorders including progressive neurological diseases such as Alzheimer’s, amyloidosis of the brain or vital organs and the induction of carcinogenesis.
From Dr McKernan’s paper, regarding DNA contamination:
Multiple assays support DNA contamination that exceeds the European Medicines Agency (EMA) 330ng/mg requirement and the FDAs 10ng/dose requirements.
…Replication competent DNA should arguably have a more stringent limit. DNA with mammalian promoters or antibiotic resistance genes may also be of more concern than just random background E.coli genomic DNA from a plasmid preparation (Sheng-Fowler et al. 2009). Background E.coli DNA was measured with qPCR and had CT over 35.
There has been a healthy debate about the capacity for SARs-CoV-2 to integrate into the human genome(Zhang et al. 2021). This work has inspired questions regarding the capacity for the mRNA vaccines to also genome integrate. Such an event would require LINE-1 driven reverse transcription of the mRNA into DNA as described by Alden et al. (Alden et al. 2022). dsDNA contamination of sequence encoding the spike protein wouldn’t require LINE-1 for Reverse Transcription and the presence of an SV40 nuclear localization signal in Pfizer’s vaccine vector would further increase the odds of integration. This work does not present evidence of genome integration but does underscore that LINE-1 activity is not required given the dsDNA levels in these vaccines.
Dr McKernan raised other concerns this week regarding the risk of plasmid related mRNA strand flipping and pathogenic protein expression:

Dr. Annelise Boucquet summarised the thread (translated):
It's even worse than I thought... If I understood correctly what @Kevin_McKernan explains, it is that the plasmid can be read in 2 directions, and the "complementary" strand (this is not quite the exact term) can encode a protein of 1252 AA of which we know nothing... When RNA is copied from DNA, if the enzyme encounters an obstacle, the reading can backfire. And they didn't put any security in place to prevent DNA reading and RNA synthesis in reverse! We therefore end up with a self-replicating DNA template that can: - either give RNA encoding the Spike - either RNA encoding parts of Spike + parts of the unknown protein - or unknown protein... To understand what's really going on, well... you have to launch the transcription experimentally and look at the nucleic acid formed. That's what I understood...it's not my field and I could be wrong. I tried to explain in simple words...
https://twitter.com/AnneliseBocquet/status/1673978920922755074?s=20
4.2.1. Drug Substance Purity
This is not the place to discuss the results of mRNA controls, but it seems important to do so solely with regard to product purity. The EMA requires a purity of 95% for products for human use [17]; according to EMA [11], the purity of the Pfizer final product is variable depending on the manufacturing process. According to the “Rapporteur Rolling Review critical assessment report” obtained by FOIA [34] (pp. 81 and 102), which details the previous document, the purity of the product is well below 95% at the time of marketing and the acceptance criterion is 50%. In another document obtained by FOIA, this threshold is 58% for mRNAs [35] (p. 38). In the 2022 report for the Moderna vaccine adapted to the Omicron strain, the EMA again asks to reassess the need to adjust the purity specification limits at the level of the active substance [46]. These defects in product purity are questionable for a new formulation.
Cumulative dose effects weren’t considered, and it could be considered that even 10 ng per dose is still too high, especially when compared like-for-like 10 nanograms is 10,000 picograms vs the pre 1986 WHO limit of only 10 picograms:
4.2.2. Drug Substance Impurities
The specification for a residual DNA template was based on the WHO recommendation: no more than a 10 ng DNA/dose [49]. This limit had been set in 1985 by the FDA at 10 pg per dose of vaccine, and raised by the WHO in 1986 to 100 pg per dose, then 10 ng per dose in 1996.
The WHO pointed out that the total number of doses to be given should be taken into account when setting this limit. Based on these considerations, and assuming a maximum dose of 30 μg, the commercial acceptance criterion at release is ≤330 ng DNA/mg RNA [33] (p. 103). However, the EMA requests further information on the linear DNA template and the quantification method.
In the EMA report [33], the results for residual DNA template and ds-RNA assays were highly heterogeneous between batches, although well below the accepted limits. It would be wise to re-evaluate assay methods and limits for future mRNA vaccines that will be evaluated outside a pandemic period. This is all the more true given that the final number of doses of COVID-19 mRNA vaccine that an individual will receive is not yet known.
The presence of contamination with a transposable element from the bacterium Escherichia coli in a batch of HPV vaccine was sufficient to get the whole production run rejected for clinical use.
Why was this cautious approach used over 10 years ago but not on this occasion, and why did it take independent researchers to find this contamination and raise awareness in the first place? The authors here recommended changing the manufacturing process and quality control methods to minimise the risk of contamination:
We report an unexpected contamination during clinical manufacture of a Human Papilomavirus (HPV) 16 E6 encoding plasmid DNA (pDNA) vaccine, with a transposon originating from the Escherichia coli DH5 host cell genome. During processing, presence of this transposable element, insertion sequence 2 (IS2) in the plasmid vector was not noticed until quality control of the bulk pDNA vaccine when results of restriction digestion, sequencing, and CGE analysis were clearly indicative for the presence of a contaminant. Due to the very low level of contamination, only an insert-specific PCR method was capable of tracing back the presence of the transposon in the source pDNA and master cell bank (MCB). Based on the presence of an uncontrolled contamination with unknown clinical relevance, the product was rejected for clinical use. In order to prevent costly rejection of clinical material, both in-process controls and quality control methods must be sensitive enough to detect such a contamination as early as possible, i.e. preferably during plasmid DNA source generation, MCB production and ultimately during upstream processing. However, as we have shown that contamination early in the process development pipeline (source pDNA, MCB) can be present below limits of detection of generally applied analytical methods, the introduction of "engineered" or transposon-free host cells seems the only 100% effective solution to avoid contamination with movable elements and should be considered when searching for a suitable host cell-vector combination.
Transposon leads to contamination of clinical pDNA vaccine, (2013).
Regarding contamination with plasmids containing the SV40 promoter, the concern with this is that if the DNA is transcripted into human DNA by chance next to a cancer promoting gene then uncontrolled cellular proliferation may result, or if mutations are induced in tumour suppressor genes then loss of function could also lead to carcinogenesis:
4. The oncogenic activity of DNA
With the identification of oncogenes in the genomes of cells [11,12], the demonstration that certain activated cellular oncogenes were able to transform primary cells in vitro to neoplastic cells [13e15], and that some of these transformed cells were able to form tumors in vivo [13,16], one scientifically based mechanism for the concern over the transfer of cancer-inducing agents from vaccine to recipient was identified, viz., through DNA capable of expressing activated oncogenes.
By analogy with viruses that integrate into the host genome, a second mechanism for DNA-induced oncogenesis has been proposed, viz., through the integration of DNA. There are several ways that the integration of DNA could have oncogenic consequences. If DNA integrated next to a dominant proto-oncogene such as c-myc and increased its expression level or activated its expression inappropriately, then this could result in the oncogenic conversion of a normal cell to one with a neoplastic phenotype. If DNA integration occurred in a tumor-suppressor gene, such as the p53 gene or the RB gene, resulting in the functional inactivation of that gene, then this cell might, over time, become neoplastic through loss of heterozygosity by acquiring additional inactivating mutations in the other allele. Both of these mechanisms have been seen following retrovirus infection in both avian and mammalian systems. For example, activation of a proto-oncogene that resulted in leukemia has been observed following infection of chickens with avian leukosis virus [17] and of mice with Moloney murine leukemia virus [18,19], and the tumor-suppressor gene p53 can be inactivated following retrovirus insertion [20,21].
Sheng-Fowler L, Lewis AM, Peden K. Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals. 2009;37(3):190-195. doi:10.1016/j.biologicals.2009.02.015
https://www.sciencedirect.com/science/article/pii/S1045105609000293
(full version is paywalled)
Antibiotic resistance genes are another major concern:
4.2.3. Problem Posed by the Presence of Antibiotic Resistance Genes
The DNA plasmid used as a template for mRNA production contains a kanamycin resistance gene [33] (p. 26). Given the significant and variable quantities of contaminating DNA in the drug substance, there is concern that the resistance gene could be integrated into human digestive tract bacteria or somatic cells [37]. If anti-COVID-19 mRNAs had been subject to GTP regulation, these studies would have been carried out.
Therefore, the controls required for all drugs and vaccines have not given completely satisfactory results concerning the product purity and quality.
And cationic lipid nanoparticle carriers (LNPs) being used in BNT162b2 and other GTPs are, even in the absence of mRNA, associated with the sustained upregulation of an interleukin associated with inflammation, autoimmune disorders and carcinogenesis - IL66.
With a half-life measured in weeks Dr. Vanessa Schmidt-Kruger expressed concerns that they persist in organs such as the liver for months or, worse still, may never be cleared completely.
Other comments from her interview on 30 January, 2021 proved to be quite prescient:
18.42
VSK: So it is theoretically possible that this linearised DNA that is in there as a contaminant could integrate into the host’s cell nucleus in a dividing cell, linearised DNA is optimal for integration. Circular DNA is not. DNA from bacteria is circular and is not as easy to integrate. It happens, but not so often. But as soon as you have a situation like we do here, it will happen more often. That is the risk. I didn’t really want to get into what can happen if this is the case: genes can be switched on and off, upregulated and downregulated, cancer can develop – there are a lot more possibilities. So this contamination definitely has to be reduced.
RF: Can you explain that to us again because that is particularly important for us as lawyers, especially for Dr. Holzeisen. What can happen in that case?
VSK: Ok. This integration: where it occurs is the nuclear genome – we can’t control that, it can happen anywhere. There are sections in the DNA that are vulnerable to it, and others that are not so vulnerable. And it is important where the DNA lands. It may land on a gene: then the gene will become dysfunctional, the protein will no longer be formed, and if it is an important protein, the cell may die, and if this continues to replicate, this can cause really massive damage. If for example it lands in an important cell that divides frequently, then clones can arise that are modified, they are gene modified, and in that case in these cloned cells these proteins are no longer produced and then, in the worst case, there is a loss of function. If it leaps into genes that have a regulatory effect on gene expression, then the genes may be switched on or downregulated, i.e., the output will differ. And this means the metabolism of the cell will alter. If this is passed on in replication, then many things may alter in the body.
WW: But these are processes that are probably not the same in all patients. Whether this happens at all is stochastic at the most, and if it does happen, the results are probably also dependent on each individual and what else is going on in their cells. So one can’t say put that in and this is what will happen, these are eventualities – if a million or so and so many thousands of people are vaccinated, then one can perhaps say with a certain degree of probability after 10 or 20 years whether something will happen or not.
VSK: Yes.
WW: With some things perhaps after 3 or 4 years. But one needs some time to be able to detect such effects clinically.
VSK: That’s exactly right. One never finds a group that all have the same mutation, this varies in people - exactly.
…And above all – this is particularly important – the cationic lipid, it is cationic, i.e. it has a positive charge. And that is very very toxic, we have known that for over 20 years.
So, that is the process for now. Do you have any questions about it?
RF: 1.06.49 That is the process after the vaccination, before you even get into the vicinity of a dangerous virus?
VSK: Yes, that’s how the immune response arises, i.e. it’s part of the immune response. It is much more extensive, there are other factors, but that is very roughly how antibodies are produced and how the antigens - the spike proteins - are destroyed in the cell.
So: I wanted to show you exactly what the toxicity is. We definitely have these cationic lipids in the cell, and now I’ll talk about what they do with the cell.
I’ll just leave the screenshare now.
Ok, that’s fine.
So: the cationic lipids. I’ll go back to the BioNTech vaccine: the LNPs consist of up to 50% of these cationic lipids: 50% is very high, they are toxic because they have this positive charge. This enables them to enter into interactions with other components of the cell really well, they can also basically interact with negatively charged amino acids. This destroys the proteins which lose their ability to function because they “unfold” as it is called. In principle they can interact with the DNA because the DNA is also negatively charged due to its phosphate groups, creating DNA strand breaks. They can also interact with other lipids because they are also negatively charged, especially the lipids of the cell membrane. E.g. the cell membrane of the mitochondria, these are the powerhouses of the cell that are vital for energy generation; I’m mentioning this because oxygen radicals are formed in the mitochondria when energy is produced. This is a very natural process, but the cell also has a repair mechanism so that these oxygen radicals are removed again and rendered harmless, this is how the cell survives, it is simply a balance. They are produced, you can’t prevent that because oxygen is consumed, which generates oxygen radicals, but avenues have been found to disable these oxygen radicals. If however these cationic lipids gain entry, it is confirmed in many publications that they destroy this membrane [Trans: meant is the mitochondrial membrane, here] and this leads to the formation of a large number of oxygen radicals. These oxygen radicals create a lot of damage in the cell. They interact – they alter the amino acids, the cell pours out as many cytokines as it can, the oxygen radicals also attack membranes and create lipid peroxidation. Membrane integrity is jeopardised, the membrane becomes porous, and when a cell membrane becomes porous water flows in and then the ion balance is disrupted. This means the entire cell loses its function because the function of proteins depends on the ion concentration, on the calcium ion for example, and the magnesium ion. The cell experiences maximum oxidative stress, as it is called in the specialist terminology. And when that stress is so high and the DNA is also damaged, then the cell goes into apoptosis – it self-destructs.
…1.16.57
If you can use the radioactivity as a marker, you can use a technique whereby can can see the organs and whether the lipids were in them or not to see. They injected the whole muscle and watched how the lipids spread out throughout the body, and found that these lipids were in many organs after just 15 minutes. Most were at the injection site, in this case it was the muscle, but a lot in the plasma, too. Logical because it’s transported in the plasma, but also 22% in the liver. And if you inject it into the veins then 60% of the cationic lipids can be found in the liver, and 20% of the PEG lipids. They were also found in the spleen, the adrenals, and in both sexual organs. Further organs were not described. So I assume that it spread out throughout all organs. 1.18.02 It is basically absorbed everywhere where blood flows. The description focuses most on the injection site, the plasma, and the liver.
Then they looked at how the lipids were degraded. They found evidence of the cationic lipid in the plasma for 12 days, and evidence of the PEG lipid for 6 days. So they remained for quite some time. There isn’t any more information, so I don’t know whether the lipids could be evidenced for longer or not. 50% of the PEG is degraded via excretion, i.e., it is excreted from the body. It goes into our “sewer system”, as it were. The cationic lipids are exclusively degraded in the cells, only 1% was found in the stool. This means the cells take the full hit of the toxicity. Then they analysed the half life of this cationic lipid in the liver, they say it is 3 weeks. With half life at the beginning the substance always degrades faster, and then it gets less, the curve gets flatter. This half life at the outset is already 3 weeks, which is relatively long. And how long does the elimination take? One can still find 5% of the lipid in the liver after 4 - 6 weeks – that is incredibly long, and with the PEG the half life is 1 week. So it is shorter, but because a large proportion, i.e., 50%, is excreted. That is not the case with the cationic lipid.
We don’t have any other information or investigations regarding other organs, they just investigated liver, plasma, urine and stool. They should definitely have looked at other organs. Perhaps they did, but there’s nothing in the publication about that.
…So to summarise, both the RNA and the LNP are taken up relatively fast. And the cationic lipids remain in our bodies for a very long time. This was also interesting. There seems to have been a discussion of the EMA with BioNTech about the period that it remains in the body: how long is it in the case of human beings, they asked, because the study wasn’t done. BioNTech referred to a study from 2010, by Mamoth et al. I have not been able to find this in the publications database, and there is no list of references below the EMA report, so I don’t know whether this is true at all and whether that article exists, but they say they have used similar lipids, and when they calculate the conversion from this mouse or rat study to human beings, that cationic lipids have a half life of 20 to 30 days in human beings, and the elimination to 5%, so not really eliminated, takes 4 - 5 months. They assume 4 - 5 months, and the EMA Committee just said “That’s a long time”. 1.22.54
Dr. H: The second vaccination comes on top of that after 30 days …
Interview with Dr. Vanessa Schmidt-Kruger
Hearing # 37 of German Corona Extra-Parliamentary Inquiry Committee
30 January, 2021
http://enformtk.u-aizu.ac.jp/howard/gcep_dr_vanessa_schmidt_krueger/

Regarding cationic damage to cell membranes and mitochondria, several post vaccination neurological disorders such as fibromyalgia (FM )may be induced or exacerbated by this, in complex with auto-immune and gp120 mediated damage and decreased blood-brain-barrier integrity, amongst others.
There is not necessarily a solid dividing line between causes and symptoms of what are medically named one condition vs another, eg FM vs peripheral neuropathy (PN) vs chronic Lyme disease (CLD) vs chronic fatigue syndrome or myalgic encephalomyelitis (ME) vs multiple sclerosis (MS).
This is why root cause analysis is important before you can think about therapeutics. The great thing about some of ours (eg baicalin7 or curcumin8) is that they can act to regulate several inflammatory or autoimmune pathways at once.
3. Mitochondrial alterations in fibromyalgia
Mitochondrial myopathies are disturbances that are characterized by morphological anomalies of the mitochondria in muscles. Mitochondrial problems are found in the most common inherited metabolic illnesses. The patients who suffer from mitochondrial myopathies can present symptomatology characterized by muscle weakness, pain, fatigue, and exercise intolerance that progressively worsen over time, similar to what happens in patients with FM [34]. Defects in any part of the cycle in the generation of ATP by the mitochondria can alter mitochondrial energy production and cause symptoms [35]. Oxidative stress is implicated in the pathogenesis of FM, which indicates that mitochondrial dysfunction can be associated with FM [36]. In fact, a decrease in the quantity of mitochondrial mass and the coenzyme Q10 (CoQ10) in the production of mitochondrial ROS in mononuclear blood cells has been detected in patients who suffer from FM [37]. Reports of muscle biopsies from the trapezius muscle have shown inflammatory markers, abnormal mitochondria, accumulation of sub-sarcolemma mitochondria, higher incidence of irregular red fibers, and defects of the cytochrome-c oxidase (Complex IV of oxidative phosphorylation) [38]. In addition, the implication of mitochondrial oxidative stress in peripheral nociception described as a predominant symptom mediated by the inflammatory state in FM has been previously reported [39].
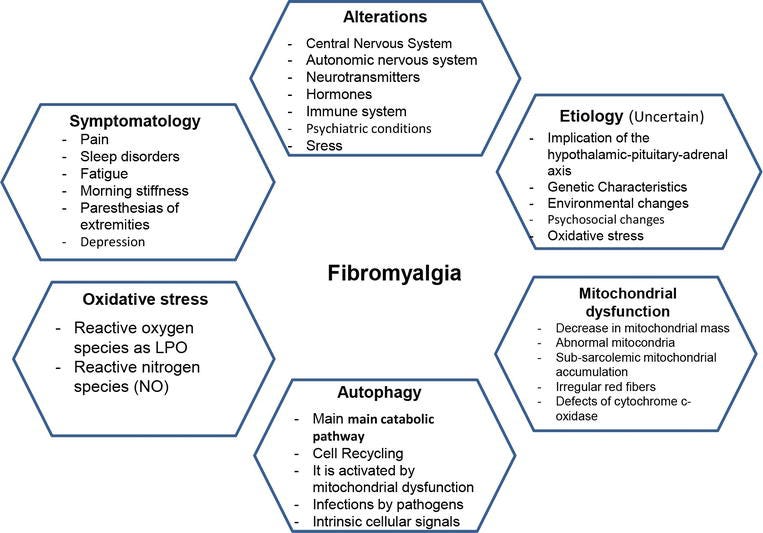
Miranda-Díaz AG, Quetzalcóatl Rodríguez-Lara S, Miranda-Díaz AG, Quetzalcóatl Rodríguez-Lara S. The Role of Oxidants/Antioxidants, Mitochondrial Dysfunction, and Autophagy in Fibromyalgia. In: Discussions of Unusual Topics in Fibromyalgia. IntechOpen; 2017. doi:10.5772/intechopen.70695
Its not only the LNPs but both GTP and viral mRNA expressed spike protein has been found in tissues 1+ years after transfection and in the skull-meninges-brain axis of those whose deaths which may have been ascribed to other causes:
Our results revealed the accumulation of the spike protein in the skull marrow, brain meninges, and brain parenchyma. The injection of the spike protein alone caused cell death in the brain, highlighting a direct effect on brain tissue. Furthermore, we observed the presence of spike protein in the skull of deceased long after their COVID-19 infection, suggesting that the spike’s persistence may contribute to long-term neurological symptoms.
Rong Z, Mai H, Kapoor S, et al. SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-Brain Axis: Potential Implications for Long-Term Neurological Complications in post-COVID-19. Published online April 5, 2023:2023.04.04.535604. doi:10.1101/2023.04.04.535604
4.3.1. Pharmacokinetics of Anti-COVID-19 mRNAs
The pharmacokinetic controls required for a new vaccine formulation have not been fully performed. It is unfortunate that complete pharmacokinetics studies have not been fully conducted since the EMA points out that several reports in the literature indicate that LNP-formulated RNAs can distribute rather non-specifically to several organs such as the spleen, heart, kidney, lungs and brain [11] (p. 54). Moreover, independent post-marketing studies have shown the distribution and persistence of the mRNA for several weeks in many organs [50,51,52]. The product of the mRNA, the spike, also circulates in the blood for several weeks [53,54,55,56]. The spike protein was found in the brain and the heart of a person who died 3 weeks after vaccination [57]. The spike was found in skin lesions up to 100 days after vaccination [58]. The preclinical studies provided by the manufacturers, therefore, appear to be incomplete from a pharmacokinetic standpoint, as they failed to detect this broad biodistribution and persistence.
As above, 5% after 4-5 months is not “complete elimination”:
For Pfizer, only the isolated components of the nanoparticles were studied regarding biodistribution. The EMA notes, from studies on similar components carried out for a GTP (the Patisiran data), that a half-life of 20–30 days in humans, and of 4–5 months for the complete elimination of lipids from nanoparticles, can be expected. Biodistribution should have been studied on the complete nanoparticle loaded with mRNA, all the more so as preclinical studies have shown biodistribution in all organs [32].
“Vaccination” with GTPs can induce spike protein levels that exceed those associated with severe COVID-19 infections. And then you may repeat the process up to 6 times via boosters, as at the time of writing:
According to Spikevax-EPAR [11], biodistribution, genotoxicity and repeat toxicity studies were performed with mRNAs encoding proteins other than the SARS-CoV-2 spike. This is not compatible with the GTP regulations, as the EMA requires that distribution studies be conducted on the transgene, as included in the GTMP [37].
These biodistribution data should have reinforced the need for certain essential GTP controls. Indeed, the EMA [38] requires that, in the event of signs of long term expression, unintended genomic integration and oncogenesis must be investigated. The duration and expression should be determined by RT-PCR and immunological assays and/or assays to detect functional protein. The over-expression of the transgene has to be monitored [38]. This should have been controlled, given that large quantities of the spike protein can be produced, sometimes in excess of those circulating in those with severe COVID-19. A comparison of spike concentrations achieved during disease and after vaccination shows that, during severe COVID-19, the median concentration observed is 50 pg/mL, with maximums at 1 ng/mL. During severe COVID-19 infection, levels of up to 135 pg/mL of S1 spike can be detected, most commonly between 6 and 50 pg/mL. After vaccination with mRNA vaccine concentrations, up to 150 pg/mL are commonly observed, but may reach 10 ng/mL in individuals with vaccine-induced thrombocytopenia [55,59].
One of the cathepsin CSL1 cleaved inserts from spike protein which shares homology with the HIV envelope protein gp120 may never be cleared from some nerve axons too:
We identified amino acid sequences in all regions of spike protein, including the S1/S2 region critical for activation and viral entry, that are susceptible to cleavage by furin and cathepsins B, K, L, S, and V using PACMANS, a computational platform that identifies and ranks preferred sites of proteolytic cleavage on substrates, and verified with molecular docking analysis and immunoblotting to determine if binding of these proteases can occur on the spike protein that were identified as possible cleavage sites.
Bollavaram K, Leeman TH, Lee MW, et al. Multiple sites on SARS‐CoV‐2 spike protein are susceptible to proteolysis by cathepsins B, K, L, S, and V. Protein Sci. 2021;30(6):1131-1143. doi:10.1002/pro.4073
DSP: Distal sensory polyneuropathy.
Since the amount of free gp120 in plasma has been determined to be in the low picomolar to low nanomolar range (Gilbert et al., 1991; Oh et al., 1992), 100 pM gp120 was perfused to determine if a lower, more physiological relevant concentration might affect transport. In fact, 100 pM gp120 impaired FAT to a similar extent as 10 nM gp120. The local concentration of gp120 around DRG neurons has not been directly measured but is likely higher than plasma concentrations due to the close proximity of infected cells binding to extracellular matrix components or local glial swelling (Nath, 2002; Krathwohl and Kaiser, 2004). An important point is that DSP takes decades to develop, although subclinical signs are commonly manifested in HIV patients. Thus, smaller amounts of gp120 internalized by DRG neurons could gradually compromise neuronal function over decades by compromising FAT in DRG neurons.
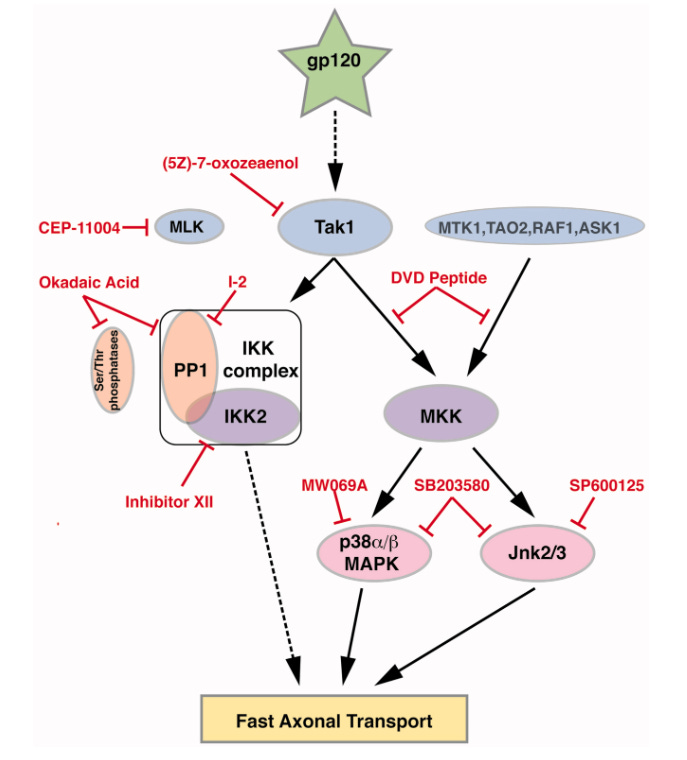
Berth SH, Mesnard-Hoaglin N, Wang B, et al. HIV Glycoprotein Gp120 Impairs Fast Axonal Transport by Activating Tak1 Signaling Pathways. ASN Neuro. 2016;8(6):1759091416679073. doi:10.1177/1759091416679073
For a deep dive:
Symptoms and clinical cases requiring extensive medical interventions are likely to be presented in increasing numbers for decades to come. Diagnosis will be challenging, root causes not directly assignable and you will be very unlikely to win compensation by legal challenge or through government run payment schemes.
You will be on your own financially and at the end of a very long and growing waiting list, made worse due to the consequences of previous vaccine mandates on the clinical care staff you will be consulting.
There are alternatives (I mentioned 2 earlier), provided the damage isn’t too extensive:
Genomic integration studies were not conducted. The consequences could be generational through offspring too, if they survive long enough and remain fertile:
4.3.2. This Broad Biodistribution Should Have Made the Carrying Out of Controls Required for GTP Essential
Germline Integration
The possibility of a vertical germline needs to be investigated (signals in gonads, signals in gametes, semen fractionation studies and integration analysis), especially since the EMA emphasizes a broader biodistribution pattern with low and measurable radioactivity in the ovaries and testes [11]. Genome integration studies are required for GTMP [24]. It was specified in 2009 that, for gene therapy medicinal products not expected to be capable of integration, integration studies must be carried out if the biodistribution of the product indicates a risk of germline transmission [13]. A 2005 document, dedicated specifically to the study of germline transmission of gene transfer vectors and naked DNA, specifies that only DNA, and not RNA, is presumed to pose a risk of germline modification [60]. This assertion can be questioned on the basis of two publications showing that, firstly, SARS-CoV-2 RNA can integrate into the genome [61] and, secondly, that the vaccine mRNA may be able to integrate into the genome of human cells in culture [62]. Although contested [63], these results would tend to require genome integration studies for mRNAs, especially since spike mRNA also translocates into the nucleus [64].
As we have seen previously the GTPs do not stay in the deltoid muscles and they knew this. If they did then they wouldn’t generate much of an immune response, just local inflammation. Its an absurd excuse to use and the regulators should have been straight on to this:
Genotoxicity
Preclinical genotoxicity studies in rats for Moderna showed equivoqual results [65] (p. 21). The conclusion was that “Overall, the genotoxic risk to humans is considered to be low due to minimal systemic exposure following IM administration, limited duration of exposure, and negative in vitro results”. It would have been wise to continue these studies, since exposure is not limited to muscle, nor is the duration of exposure. As specified in the EMA regulations for GTPs “If a positive finding occurs, additional testing will be needed to ensure the safety of the product” [38].
Teratogenic: Of, relating to, malformations or defects to an embryo or foetus.
This should have at least resulted in restricting licensing to non-pregnancy, if not a complete halt to all trials pending further investigations:
Reproductive and Developmental Toxicity
According to a document concerning non-clinical trials of the Moderna vaccine obtained by the FOIA, skeletal variations (one or more rib nodules and one or more wavy ribs with no effect on the viability or growth and development of F1 generation) appeared in the fetuses of vaccinated rats but were not considered adverse. However, it is emphasized that they appeared at the same time as maternal toxicity correlating with the most sensitive period for rib development in rats [65].
Nothing is ever considered to be “treatment related”, yet they cannot offer alternative explanations and never extended the studies:
According to documents obtained by the FOIA from Australian [66] and Japanese [67] regulatory agencies, skeletal malformations were also found in the Pfizer preclinical trial. The incidence of supernumerary lumbar ribs was higher in the treatment group compared with the control group, but was not considered to be treatment-related. This concordance of fetal anomalies with the two types of mRNA vaccines should have led to more detailed studies.
Regarding pharmacokinetics:
Extended dosing should have resulted in further safety studies, as referred to earlier.
All the components should have been considered as potentially biologically active ingredients and tested as a whole unit, not just individually.
You can’t just submit one product for approval and then alter it or change administration guidance later without consequence to your licensing (at least in a world where safety isn’t just a profit-killing inconvenience).
2 years to assess for carcinogenicity is an extremely short period, but it would have been an improvement on 42 days.
Interactions with long noncoding RNAs (lncRNAs) and oncogenic miRNAs were never assessed, which has implications for carcinogenesis, viral latency and impaired immune responses:
For a new excipient, the chemical, pharmaceutical and biological information should be identical to that provided for the active substance [17] (p. 135). Furthermore, according to Hemmrich and McNeil [68], the status of LNP components is confused, according to the FDA; they are considered either as “starting materials” (and therefore not as excipients) or as “inactive ingredients” (and therefore excipients) according to the documents whereas, according to the FDA itself, they should be considered as active ingredients. According to the same authors, developers must demonstrate the safety of the new ingredient, and “excipients intended for long-term use may require repeated dose toxicology studies over 6 months and carcinogenicity studies over 2 years”. Indeed, the manufacturers intended only two doses of vaccine [46], but some populations are currently receiving up to six doses spaced a few months apart. The stability, toxicity and biodistribution of the intact nanoparticle containing the mRNA, the active substance, must be evaluated, rather than the isolated lipid components, contrary to what has been carried out (Moderna and Pfizer have partially evaluated the biodistribution of lipids in nanoparticles, or of nanoparticles containing mRNAs other than those used in anti-COVID-19 vaccines) [11,32,33,34].
The FDA classification of these mRNAs as GTPs would have resolved these ambiguities since the FDA recommends assessing the risks of the GTP delivery procedure (biodistribution in blood, cerebrospinal fluid, germline, heart and brain to be assessed in preclinical trials, persistence of the vector). The FDA also requires the evaluation of potential horizontal transmission of the replication-competent vectors from the patient to family members and health care providers (i.e., shedding). This requirement should have applied even if the vector is not a replication-competent virus.
As for clinical studies, despite licensing for use in pregnancy and being experimental in nature no data were available on vaccine placental transfer or excretion in milk:
4.4. Clinical Studies
As shown above, the spike protein has been shown to circulate well in the blood, excretion studies should therefore have been carried out. The CHMP noted that no data are available on vaccine placental transfer or excretion in milk [11] (p. 56). Studies independent of the manufacturers have shown the passage of vaccine mRNA into breast milk in the first week following injection [69,70,71,72] and adverse effects on breast-fed babies could be due to this passage, according a FDA report [73]. Nanoparticles, similar to those in COVID-19 mRNA vaccines, have been shown to be able to cross the placental barrier in mice [74]. Extensive preclinical and clinical studies should have explored this passage in milk and through the placenta.
Information on carcinogenicity was a black hole, despite knowledge of cancer pathways that viruses, LNPs and GTP mRNAs can signal.

Immunosuppression and igG4 class switching data should also have been gathered over extended periods and emergency licenses withdrawn as the research findings were published.
A prophylactic medication must never be more pathogenic and carry a higher fatality rate than the disease you are trying to prevent, especially in lower risk cohorts (ie all those apart from the elderly with multiple comorbidities and even then, due to immunosuppression and escape variants they would have been better served by taking antivirals).
Most of my earlier Substacks focused on these areas.
Carcinogenicity, tumorigenicity and immune suppression studies should have been carried out, because two studies have suggested that mRNA vaccines may induce immunotolerance [75,76]. In addition, the spike protein may interact with the tumor suppressor [77,78] (p. 53). It would therefore be wise to explore the tumorigenic effect in vivo and to monitor any cancers developed by vaccinated individuals over the long term, especially as it has been suggested that cancers can be reactivated by mRNA vaccines [79,80,81] or may develop after mRNA vaccination [82,83,84,85,86].
In practice, few cases appear to have been followed up long term and if they did collect any useful data they are not making it public. Now why would that be?

They also destroyed the placebo group (perhaps literally in some cases) by transfecting them shortly after the trial. This shouldn’t stop them from conducting retrospective studies to compare rates of adverse events with the unvaccinated. Its all very convenient for hiding the bodies:
4.5. Vaccinovigilance
GTP regulations require the very long-term monitoring of adverse effects. This will be difficult to achieve for mRNA vaccines because the EMA has requested a 24-month follow-up of adverse events after vaccination, pointing out that a significant number of participants in the placebo group were vaccinated, which makes this follow-up more difficult [34] (pp. 14, 114 and 138). Moderna announced that “as of 13 April 2021 all placebo participants have been offered the Moderna COVID-19 vaccine and 98% of those have received the vaccine” [87].
The latest date for pharmacovigilance follow-up required by the EMA is 31 March 2024 [11], well below the FDA’s long-term follow-up of adverse events for GTP, which is 5 to 15 years, and 30 years for the EMA. In addition to the extensive pharmacovigilance plan requested by the EMA [11], we could call for reinforced monitoring. As the placebo groups in the clinical trials were vaccinated, the long-term monitoring of these adverse effects could be carried out by retrospective observational studies comparing the incidence of pathologies according to participants’ vaccination status. Two publications about adenovirus vectors [88,89] suggest that the long-term effects of gene therapy vectors, although they are not specifically mRNA vectors, clearly exemplify the lack of study and potential risks in the long term.
The FDA and EMA recommend the long-term monitoring of possible adverse effects of GTPs, particularly for certain diseases (cancers, hematological, neurological, rheumatological conditions and infections). We have seen above that there are reported cases of new or reactivated cancers following anti-COVID-19 mRNA vaccination. There are also reports of cases of diseases specifically to be monitored after GTP administration. Here are just a few examples, as it is impossible to be exhaustive. Concerning hematological disorders, there have been reports of bone marrow suppression [90] and aplastic anemia [91]. For neurological conditions, encephalitis [57], rhomboencephalitis [92], demyelinization [93] and autoimmune neurological diseases [94] were noted. Rheumatological conditions [95], de novo autoimmune rheumatic diseases [96], autoimmune like myopathy [97] and new or exacerbated inflammatory diseases [94] have also been reported.
A recrudescence of brain abscesses in 2021, after the start of the massive vaccination campaign, is reported [98] and non-covidial pneumonias following mRNA vaccines [99].
The EMA points out that an interventional study is underway to assess the safety and tolerability of Pfizer’s vaccine in pregnant women [11]; although the actual study completion date is 15 July 2022, no results have been published [100], which is unfortunate.
Yes, there is a statistical correlation between brain abscesses and later cancer incidence:
Results Among 1,384 patients with brain abscess (37% female, median age 50 years, interquartile ranges [IQR] 33–63), cancer was observed in 218 (16%) compared with 1,657 of 13,838 (12%) in the comparison cohort yielding an adj. HRR of 2.09 (95% CI 1.79–2.45). The median time to diagnosis of cancer was 1.8 years (IQR 0.02–9.1) in patients with brain abscess and 8.6 years (IQR 3.9–15.9) in comparison cohort. Among patients with brain abscess, CNS and eye cancer was diagnosed in 59 (4.3%), of which 47 of 59 (80%) occurred within 90 days of the admission date, metastasizing cancer in 54 (3.9%), respiratory tract cancer in 48 (3.5%), and gastrointestinal cancer in 36 (2.6%). Results remained consistent in almost all subgroups and in sensitivity analyses.
…Follow up of over 1000 patients treated for cerebral abscess revealed that the incidence of cancer was doubled during the first 10 years after diagnosis in comparison with a control group matched for age, sex, and place of residence.
Bodilsen J, Søgaard KK, Nielsen H, Omland LH. Brain Abscess and Risk of Cancer: A Nationwide Population-Based Cohort Study. Neurology. 2022;99(8):e835-e842. doi:10.1212/WNL.0000000000200769
And to conclude the review as well as this walkthrough we have their conclusions, reproduced here in full:
5. Conclusions
Although the principle of action of COVID-19 mRNA vaccines corresponds to the definition of gene therapy products (GTPs), they have been excluded from the regulation of GTPs by the regulatory agencies (US-FDA and EMA) and subjected to the regulation of vaccines against infectious diseases. No scientific or ethical justification is given for this exclusion, and there remain inconsistencies in the regulations. For example, under European and French regulations, a vaccine must contain an antigen, which is not the case for mRNA vaccines. These products could be considered “pro-vaccine”. In fact, mRNA vaccines do not contain an antigen, but make the vaccinee produce it. They can therefore be classed as pro-drugs or “pro-vaccine”. Special regulations should be drawn up for this type of product, insisting on potency controls, i.e., the quality, quantity, duration and sites of expression of the antigen of interest, as well as the toxicity of this antigen. As proposed at the start of 2020, the SARS-CoV-2 spike protein interacts with the renin-angiotensin system [101,102,103] and has a recognized toxicity that was known since before COVID-19 [104] and has been confirmed since [105,106,107,108].
According to European regulations, vaccines are human medicinal products and must therefore undergo the same controls, but not all of these controls are generally applied to vaccines against infectious diseases. With regard to the controls applied to mRNAs, it is worth noting that the degree of purity of the product is lower than that required for any drug: this is questionable for a new formulation and principle of action. It is also possible that batch heterogeneity was not detected by the batch release procedure. Impurities linked to this new formulation could pose safety problems; the presence and quantity of contaminating DNA from the template used to manufacture the RNA and of ds-RNA would need to be reassessed. The presence of antibiotic resistance genes in contaminating template DNA also raises safety issues.
Pharmacokinetic studies are not generally required for vaccines, except in the case of new formulations, which is the case here. However, extensive studies in this field would have been necessary, since they did not detect the wide distribution and persistence of mRNA, and its product, the spike protein, in the bodies of vaccinees, the passage of mRNA in breast milk, nor the possible passage through the placenta of vaccinated mothers. GTP regulations require these in-depth studies on the complete formulation (the lipid nanoparticle loaded with the mRNA corresponding to the drug product).
Because of this wide and persistent biodistribution, essential tests required for GTPs should have been carried out regarding: the risk of genotoxicity, genome integration and germ-line transmission, insertional mutagenesis, tumorigenicity, embryo/fetal and perinatal toxicity, long term expression, repeated toxicity and excretion in the environment (shedding in the seminal fluid, for example).
The long-term safety monitoring of GTPs is required over several years whereas, for vaccines, it is generally only carried out over a few weeks. This should not be acceptable, given the persistence of the drug product and the expressed protein. The known results of anti-cancer therapies and mRNA vaccines could lead us to anticipate problems of safety and efficacy. In the case of anti-cancer mRNAs, the vast majority of open-label clinical trials have been carried out on very small numbers of patients, with either unpublished or negative results [109,110]. Randomized studies also showed negative results, reporting more frequent adverse events in the treatment group [111,112]. Concerning infectious diseases, two trials of mRNA vaccines encapsulated in LNPs showed notable adverse effects. A trial of an mRNA vaccine against rabies showed numerous adverse effects superior to those of the classic vaccine, which is already very reactogenic, notably lymphopenia (this effect was also found for anti-COVID-19 mRNA vaccines) [113]. An influenza vaccine trial [114] showed severe adverse effects in humans (31 subjects were observed over only 43 days and at least 4 serious adverse effects were found). In a non-randomized trial against HIV [115], the response was inexplicably incomplete in some patients. According to another HIV trial of 15 participants against a placebo, immune responses were unsatisfactory and of limited duration [116]. The founder of BioNTech himself, Ugur Sahin, warned against the use of codon optimization, which can alter translation speed and lead to misfolding. He also underlined the potential toxicity of unnatural nucleotides. He also mentioned the wide biodistribution of mRNA injected intramuscularly. He reminded us that we should fear the appearance of anti-self mRNA antibodies in patients suffering from autoimmune diseases [27].
The role of regulatory agencies is to ensure the safety and efficacy of medicines. The COVID-19 pandemic emergency has accelerated the timetable for the production and clinical use of COVID-19 vaccines; it is, therefore, possible that certain safety aspects have not been fully addressed. It is, therefore, important to take these aspects into account in the future, so as not to undermine public confidence in vaccines in general.
The WHO declared an end to the emergency phase of the COVID-19 pandemic at the beginning of May 2023 but will continue to authorize the use of the Emergency Use Listed (EUL) procedure. The emergency authorization of vaccines should be transformed into prequalification via a smooth transition [117]. However, a wide-ranging public discussion should be opened on this transition to the routine use of mRNA vaccines, without them being subject to the controls required for GTPs.
In the EMA document designed to regulate the clinical evaluation of new vaccines from 2023, there is no mention of mRNA vaccines, and it is still specified that vaccines contain antigens; this document would therefore not apply to mRNA vaccines that do not contain antigens. It is once again specified that nonclinical pharmacokinetic studies might be applicable when new delivery systems are employed or when the vaccine contains novel adjuvants or excipients. It is a pity that these points have not been specified specifically for mRNA vaccines [118]. An article from early 2021 [119] emphasized the need for further studies to ensure the quality, efficacy and safety of mRNA vaccines; it was written before these products were marketed. It seems important to clarify which additional controls should be required in light of the detailed results of the preclinical trials and safety data published in the post-marketing phase.
In the future, it should be discussed whether all mRNA-based products should be subject to the same regulations and controls, whether or not they are considered vaccines. It is not justifiable to subject therapeutic mRNAs to strict controls when they are intended for patients representing a small proportion of the human population, and to exclude from these controls mRNA vaccines intended for the majority of the healthy human population.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The publication fees were paid by aimsib.org (accessed on 1 June 2023).
Conflicts of Interest
The author declares no conflict of interest.

Disclaimer
This site is strictly an information website about potential therapeutic agents and a review of associated research papers. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
References
Banoun H. mRNA: Vaccine or Gene Therapy? The Safety Regulatory Issues. International Journal of Molecular Sciences. 2023;24(13):10514. doi:10.3390/ijms241310514
Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. Journal of Hematology & Oncology. 2022;15(1):28. doi:10.1186/s13045-022-01247-x
https://jhoonline.biomedcentral.com/articles/10.1186/s13045-022-01247-x
McKernan, Kevin & Helbert, Yvonne & Kane, Liam & McLaughlin, Stephen. (2023). Sequencing of bivalent Moderna and Pfizer mRNA vaccines reveals nanogram to microgram quantities of expression vector dsDNA per dose. 10.31219/osf.io/b9t7m.
Blánaid Hicks, James A. Kaye, Laurent Azoulay, Kasper Bruun Kristensen, Laurel A. Habel, Anton Pottegård, The application of lag times in cancer pharmacoepidemiology: a narrative review, Annals of Epidemiology, Volume 84, 2023, Pages 25-32, ISSN 1047-2797, https://doi.org/10.1016/j.annepidem.2023.05.004. https://www.sciencedirect.com/science/article/pii/S104727972300090X
BREAKFAST CEREALS - ÖKOTEST STUDY POINTS OUT RISKS, 12th April 2021.
Parhiz H, Brenner JS, Patel PN, et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J Control Release. 2022;344:50-61. doi:10.1016/j.jconrel.2021.12.027
Hu Z, Guan Y, Hu W, Xu Z, Ishfaq M. An overview of pharmacological activities of baicalin and its aglycone baicalein: New insights into molecular mechanisms and signaling pathways. Iran J Basic Med Sci. 2022;25(1):14-26. doi:10.22038/IJBMS.2022.60380.13381
Jacob A, Wu R, Zhou M, Wang P. Mechanism of the Anti-inflammatory Effect of Curcumin: PPAR-γ Activation. PPAR Res. 2007;2007:89369. doi:10.1155/2007/89369
In concluding section:
"It is, therefore, important to take these aspects into account in the future, so as not to undermine public confidence in vaccines in general."
Far, far too late!! That horse is out of the barn and into the next hemisphere!
thanks for the sobering analysis ... more vodka will help disinfect me :)