Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
This story first broke in February 2022 and well publicised at the time but I hadn’t considered reviewing it too until prompted by Dr @LadyBug in the last week, who asked me what the biological consequences could be, especially given the recent cancer trends data?
Antisense is the non-coding DNA strand of a gene. In a cell, antisense DNA serves as the template for producing messenger RNA (mRNA), which directs the synthesis of a protein1.
Each nucleotide in a double stranded DNA molecule is paired with its Watson-Crick counterpart. This counterpart is called its complementary nucleotide. Double stranded DNA sequences are represented by the upper (sense) strand sequence going in the direction from its 5′- to 3′-end. The complementary sequence is thus the sequence of the lower (antisense) strand in the same direction as the upper strand. The reverse sequence is the sequence of the upper strand in the direction from its 3′- to its 5′-end. The reverse complement sequence is the sequence of the lower strand in the direction of its 5′- to its 3′-end.
MutSβ initiates DNA mismatch repair by binding to a mismatch and then forming a further complex with MutLαheterodimer4:
Current models of MMR initiation. a The translocation model suggests that an α-like loop structure forms as a result of the “bidirectional” translocation of MutS homologs when searching for the strand discrimination signal.11 b The molecular switch model postulates that MSH homologs bind to the mismatch and then slide away from the site to search for the strand discrimination signal in an ATP-dependent manner.14 c The transactivation model suggests that the MMR initiation complex remains bound to the mismatch and activates downstream nuclease activities at the strand signal via DNA bending/looping.19,20,22 d The multi-MLH loading model suggests that mismatch-bound MutS homologs recruit multiple molecules of MutL homologs flanking the mismatch.21 https://www.nature.com/articles/s41422-021-00468-y
DNA mismatch repair or MMR is a highly conserved biological pathway. It plays a key role in maintaining genomic stability and is primarily to repair base-base mismatches and insertion/deletion mispairs generated during replication and recombination5.
The SARS-CoV-2 FCS connection
In 2022, Ambati et al published their findings after conducting a BLAST search of the SARS-CoV-2 genome6.
BLAST is short for basic local alignment search tool and is an algorithm that allows researchers to compare primary biological sequences (eg for nucleotides of RNA, DNA or amino acid sequences of proteins) with those in a reference library or database. The New York Times called it the Google of biological research7.
Arkmedic posted an excellent guide to using BLAST to expose the man-made origins of COVID-19:
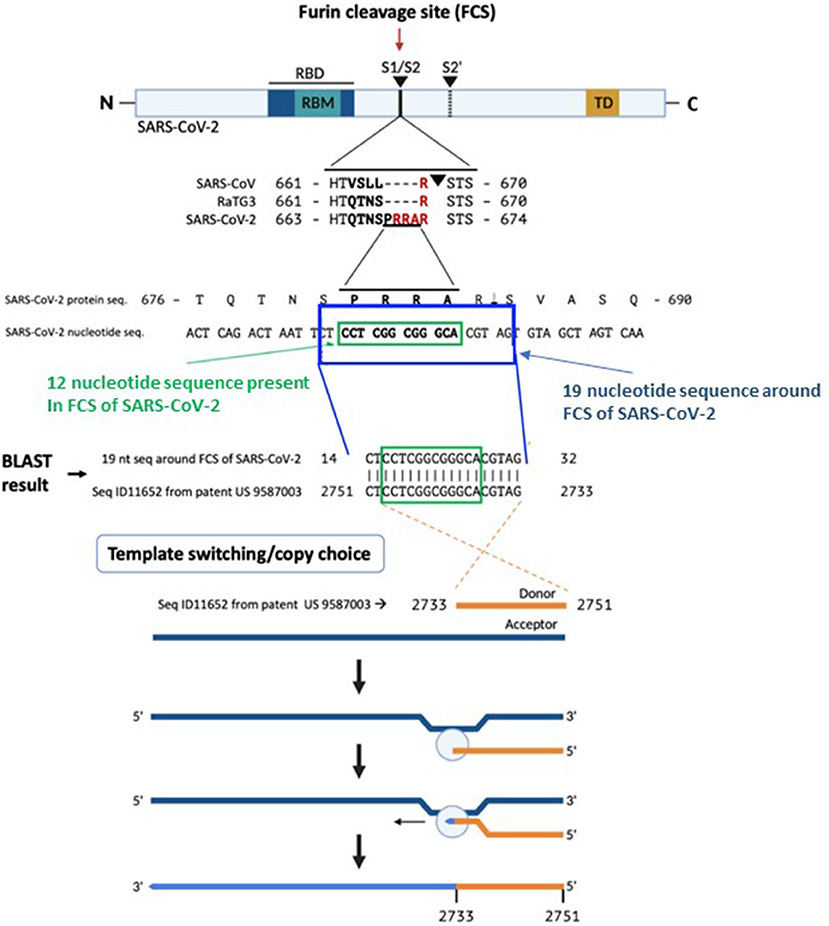
The furin cleavage site or FCS on spike S1/S2 enables cleavage by a proteolytic enzyme called furin. It also assists with escorting mRNA to the nucleus8, increasing associated pathologies (eg cell-to-cell fusion and syncytium formation which increases virulence and p53 & BRCA1 degradation, leading to failed DNA damage response9 and T cell depletion and carcinogenesis10) but it’s sequence PRRAR is “strikingly” missing in SARS-CoV1 and analogous motif R-x-x-R is insufficient to cause cleavage in MERS11.
Inclusion of the FCS leads to the release of high levels of free S1 subunit which induce further systemic pathologies. S1 induces activation of toll like receptor 4 (TLR4), which is an important component of SARS-CoV-2 induced COVID-19 disease12. It can also cross the blood brain barrier13, so questions are asked as to why it was retained in experimental mRNA gene agents despite being excluded from “vaccines” on safety grounds prior to 2020?
Key takes from their study:
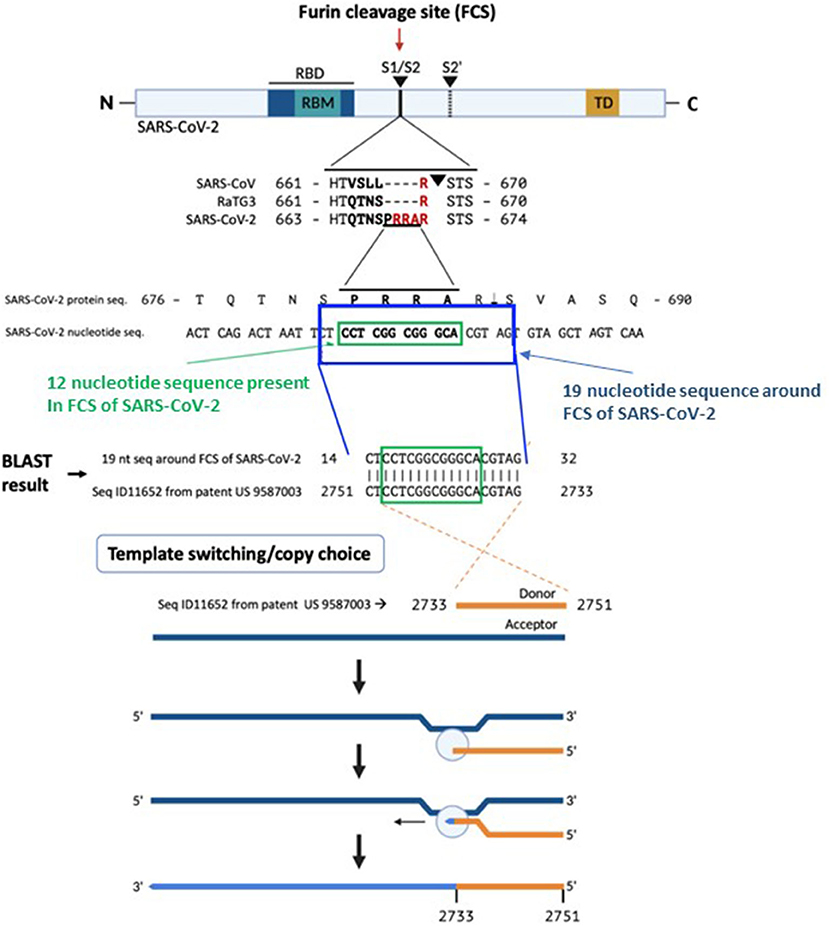
A BLAST search revealed that a 19 nucleotide portion of the genome encompassing the FCS is a 100% complementary match to a codon-optimized proprietary sequence that is the reverse complement of the human mutS homolog (MSH3).
The reverse complement sequence present in SARS-CoV-2 may occur randomly but other possibilities must be considered. Recombination in an intermediate host is an unlikely explanation.
…the presence of the 19-nucleotide long RNA sequence including the FCS with 100% identity to the reverse complement of the MSH3 mRNA is highly unusual and requires further investigations.
This polybasic FCS differentiates SARS-CoV-2 from other b-lineage betacoronaviruses or any other sarbecovirus (3). An FCS addition enhanced the infectivity of SARS-CoV in 2009 (4). The absence of this FCS results in attenuated SARS-CoV-2 variants useful for animal vaccination, accentuating its relevance to human infection (5). This FCS is vital for human and ferret transmission (6), expands viral tropism to human cells (7), and is requisite for severe disease in two animal models of SARS-CoV-2 (5).
The correlation between this SARS-CoV-2 sequence and the reverse complement of a proprietary mRNA sequence is of uncertain origin. Conventional biostatistical analysis indicates that the probability of this sequence randomly being present in a 30,000-nucleotide viral genome is 3.21 ×10−11.
The proprietary sequence SEQ ID11652, read in the forward direction, encodes a 100% amino acid match to the human mut S homolog 3 (MSH3) (9, 10). MSH3 is a DNA mismatch repair protein (part of the MutS beta complex) (10). SEQ ID11652 is transcribed to a MSH3 mRNA that appears to be codon optimized for humans (11). We did not find the 19-nucleotide sequence CTCCTCGGCGGGCACGTAG in any mammalian or viral genomes except SARS-CoV-2 with 100% coverage and identity in the BLAST database.
MSH3 replacement with a codon-optimized mRNA sequence for human expression likely has applications in cancers with mismatch repair deficiencies. While a portion of a reverse complement sequence being present in SARS-CoV-2 could be a random coincidence, other possibilities merit consideration.
Overexpression of MSH3 is known to interfere with mismatch repair (MSH2 sequestration from the MutS alpha complex comprising MSH2 and MSH6 results in MSH6 degradation and MutS alpha depletion) (12), which holds virologic importance. Induction of DNA mismatch repair deficiency results in permissiveness of influenza A virus (IAV) infection of human respiratory cells and increased pathogenicity (13). Mismatch repair deficiency may extend shedding of SARS-CoV-2 (14).
The absence of CTCCTCGGCGGGCACGTAG from any mammalian or viral genome in the BLAST database makes recombination in an intermediate host an unlikely explanation for its presence in SARS-CoV-2. A human-codon-optimized mRNA encoding a protein 100% homologous to human MSH3 could, during the course of viral research, inadvertently or intentionally induce mismatch repair deficiency in a human cell line, which would increase susceptibility to SARS-like viral infection.
The presence in SARS-CoV-2 of a 19-nucleotide RNA sequence encoding an FCS at amino acid 681 of its spike protein with 100% identity to the reverse complement of a proprietary MSH3 mRNA sequence is highly unusual. Potential explanations for this correlation should be further investigated.
The authors do not directly refer to gain of function laboratory research or biological weapons research by state actors but the inference is there that its presence is unlikely to be by chance alone of natural occurrence and is of long established known pathological significance.
Most revealing is their reference to a patent filed by Moderna in 20121415:
Based on a BLAST search of the 12-nucleotide stretch coding for the FCS PRRA, a 19-nucleotide long identical sequence was identified in the patented (US 958 7003) sequence Seq ID11652. SEQ ID11652 is transcribed to a MSH3 mRNA that appears to be codon optimized for humans. This 19-nucleotide sequence including 12 nucleotides coding for the FCS PRRA, present in the human MSH3 gene might have been introduced into the SARS-CoV-2 genome by the illustrated copy choice recombination mechanism in SARS-CoV-2 infected human cells overexpressing the MSH3 gene.
Pathologies induced by over-expression do not appear to have been a consideration of the “invention”. Key takes from the full patent application:
With the host of undesired consequences brought about by Standard treatments such as chemotherapy and radiotherapy used today, genetic therapy for the manipulation of disease-related peptides provides a more targeted approach to disease diagnosis, treatment and management. To this end, the inventors have shown that certain modified mRNA sequences have the potential as therapeutics with benefits beyond just evading, avoiding or diminishing the immune response.
The present invention addresses this need by providing nucleic acid based compounds or polynucleotide-encoding nucleic acid-based compounds (e.g., modified mRNA or mmRNA) which encode an oncology-related polypeptide of interest and which have structural and/or chemical features that avoid one or more of the problems in the art.
The present invention provides a method of treating a disease, disorder and/or condition in a Subject in need thereof by increasing the level of an oncology-related poly peptide of interest comprising administering to said subject an isolated polynucleotide encoding said oncology-related polypeptide.
To avoid breaching another patent, they can file to sequence a FCS in their “invention”, provided the polypeptide used isn’t glucagon-like peptide 1 (GLP-1).
GLP-1 may be familiar to you, its related to the weight loss drug “Ozempic” (semaglutide) and diabetes drugs, a GLP-1 agonist (promoter)16.
In one embodiment, the oncology-related polypeptides of the present invention include at least one protein cleavage signal and/or site. As a non-limiting example, U.S. Pat. No. 7,374,930 and U.S. Pub. No. 20090227660, herein incorporated by reference in their entireties, use a furin cleavage site to cleave the N-terminal methionine of GLP-1 in the expression product from the Golgi apparatus of the cells. In one embodiment, the polypeptides of the present invention include at least one protein cleavage signal and/or site with the proviso that the polypeptide is not GLP-1.
It maybe by pure coincidence or chance that the same 19nt long sequence is patented, that furin cleavage sites are specified in the same patent and also appear in both mRNA gene tech and a virus some 7 years later. I will let the reader decide on the likelihood of this?
We will explore pathologies associated with MSH3 overexpression and mismatch repair failure in the following sections.
Related Pathologies
Presence of the FCS in the viral genome was confirmed by the BLAST search as discussed previously, but is it also in synthetic mRNA gene agents too?
Fortunately this is quite simple to check for yourself, don’t take my word for it!
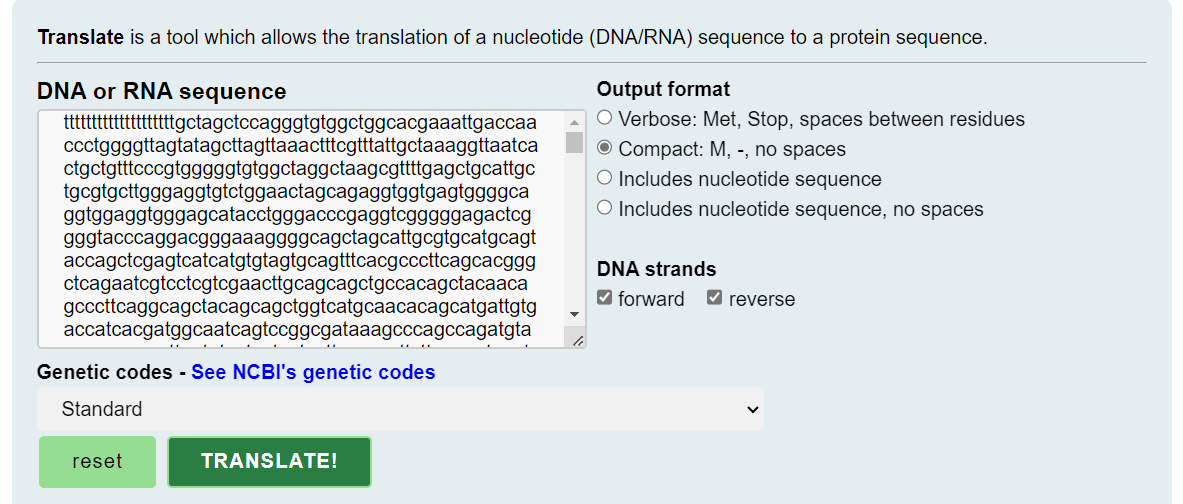
Dr. Kevin McKernan linked to and reproduces the RNA sequence for Pfizer’s BNT162b2 in his Substack17. This is part of it, you can copy the full sequence to your clipboard from his post:
Paste it into a translate tool, which allows the translation of a nucleotide (DNA/RNA) sequence to a protein sequence. This is effectively simulating simple protein expression. You can use the default settings18:
Use your browser’s search tool to look for the amino acid sequence for furin, QTNSPRRAR and sure enough, there it is:
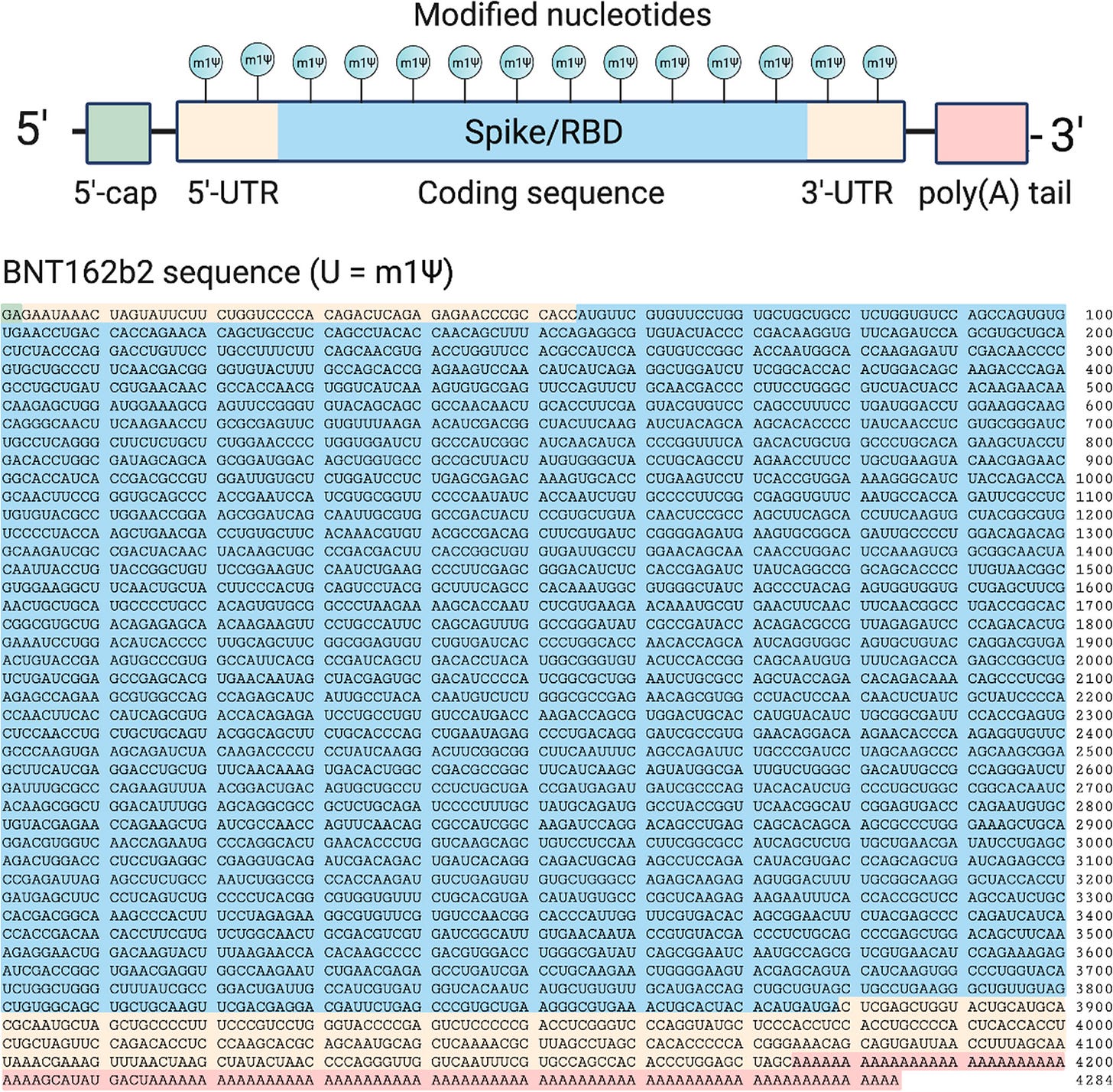
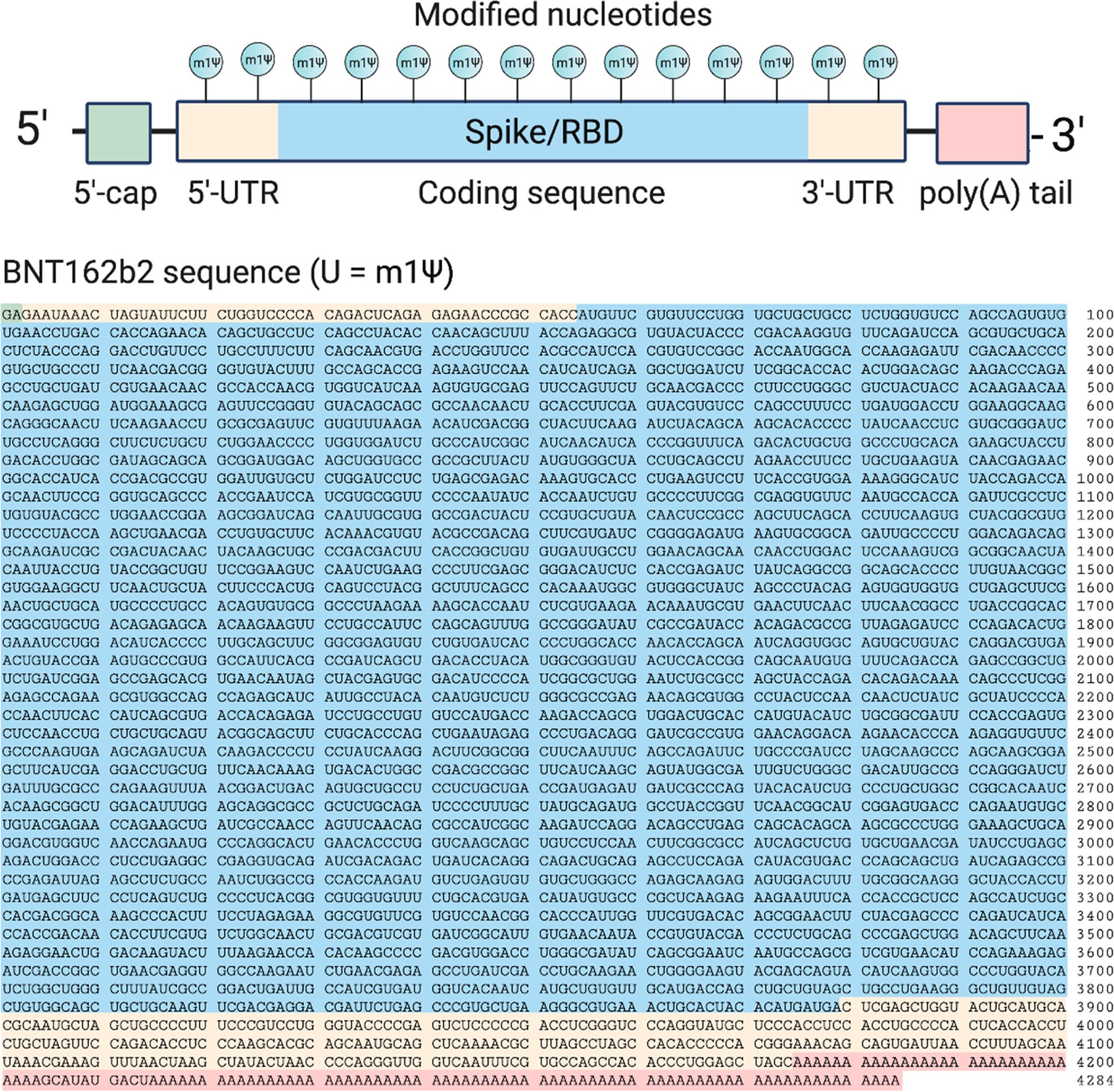
The commercial version of this has the Thymidines (T) replaced by Uridines (U), which are later converted into non-natural RNA nucleobase N1-methylpseudouridines (m1Ψ) during in vitro transcription, including for the FCS19:
Top: Design elements found in synthetic mRNA therapeutics. Bottom: Sequence of the COVID-19 mRNA vaccine tozinameran (BNT162b2) from Pfizer/BioNTech. Green: 5′-cap. Yellow: 5′- and 3′-UTR sequences. Blue: SARS-CoV-2 spike glycoprotein coding sequence. Red: Segmented poly(A) tail. https://pubs.acs.org/cms/10.1021/acscentsci.1c00197/asset/images/large/oc1c00197_0002.jpeg
Methyl pseudouridylation (m1Ψ) is used to attenuate immunostimulatory potential20 and increase protein production from synthetic mRNAs21.
Production of m1Ψ mRNAs by in vitro transcription. Left: Components of in vitro transcription reaction. Right: Incorporation of m1Ψ-triphosphate into RNA is guided by m1Ψ’s ability to form a canonical base pair with adenine of the DNA template in the T7 RNA polymerase active site. https://pubs.acs.org/cms/10.1021/acscentsci.1c00197/asset/images/large/oc1c00197_0003.jpeg
In the case of oncogenic microRNA-21 (miRNA-21), methyl pseudouridylation was found to increase protein expression multi-fold vs native type22:
Scheme and fold-change of mRNA circuits carrying different base modifications in 293FT cells.miR-21-5p mimic was co-transfected to induce ON state of the circuits. Error bars indicate the mean ± standard error (n = 3).Note that the Y-axis is exponential. Extracted from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7102939/
Another patent from 2006/2012 (filed by the University of Pennsylvania) is also enlightening as it confirms what was known years ago23.
Lipofectin® forms lysosomes with mRNA or DNA and is used for transfection, particularly into endothelial cells24.
...In expression studies provided herein, translation was measured from RNA complexed to Lipofectin® (Gibco BRL, Gaithersburg, Md., USA) and injected into the tail vein of mice. In the spleen lysates, pseudouridine-modified RNA was translated significantly more efficiently than unmodified RNA (FIG. 12B). Under the conditions utilized herein, efficiency of transfection-based methods of the present invention correlates with the ability of the transfection reagent to penetrate into tissues, providing an explanation for why the effect was most pronounced in spleen cells. Splenic blood flow is an open system, with blood contents directly contacting red and white pulp elements including lymphoid cells.
If you need to generate as much spike protein for as long as possible from your synthetic mRNA then this is ideal, but you will also, by implication, be expressing MSH3 efficiently too in a 1:1 ratio to spike protein. And there is experimental and autopsy evidence that this process may continue for months or even years, with or without viral mRNA25.
Indeed, 30% of patients with long COVID or post-acute sequelae of COVID-19 (PASC) had evidence of persistent viral mRNA and spike protein up to 1 year or longer after acute SARS-CoV-2 infection26. Vaccine boosters or breakthrough infections will further add to this load.
Cancers
Marra et al (1998) discussed how mismatch repair deficiency can arise not only through mutation or transcriptional silencing of a mismatch repair gene, but also as a result of imbalance in the relative amounts of the MSH3 and MSH6 proteins due to overexpression27.
Key takes from their study:
Western blot experiments indicate that extracts ofmammalian cells in culture generally contain only very low amounts of MSH3, despite the fact that the MSH3 gene is expressed and its mRNA can be shown to be present in a quantity similar to that of the DHFR message (16, 27).
Under normal growth conditions, all three MutS homologues are expressed, but MSH2 forms a heterodimer preferentially with MSH6, because both of these proteins are expressed very efficiently. Any MSH3 that may be translated either fails to find available free MSH2, or it becomes degraded in the absence of its cognate partner. In the case where MSH3 is overexpressed, such as in the lines described above, the situation is reversed: MSH2 complexes predominantly with MSH3, and MSH6 is degraded (Fig. (Fig.4;4; see also ref. 10).
There are implications to this concerning relapses from chemotherapy for cancer:
MTX is highly effective in the treatment of childhood acute lymphocytic leukemia, but it is also used routinely in the clinic to treat other tumors, such as osteosarcoma and breast cancer.
Resistance to MTX caused by amplification of the DHFR gene is frequently encountered in the clinic (30), and it is possible that even a small increase in hMSH3 levels could significantly alter the relative intracellular concentrations of hMutSα and hMutSβ and thus lower mismatch repair efficiency. Importantly, because amplification of the DHFR gene under MTX-selective pressure was reported to be exacerbated by induction of expression of the c-myc oncogene (31), which is overexpressed frequently in tumors, it is conceivable that the amplification process might be facilitated in cancer. This hypothesis is supported by numerous reports showing that increased expression (whether through genomic amplification or transcriptional activation) of genes involved in drug resistance (including DHFR) appears to be common in tumors lacking the product of the Rb gene, overproducing cyclin D1, or with altered p53 (see ref. 30 for review). Because p53 mutations are found in around 50% of all cancers (32), it is possible that MTX therapy might select for clones with diminished mismatch repair efficiency whose mutator phenotype would accelerate the acquisition of mutations in other tumor suppressor genes and oncogenes that are necessary for the progression of malignant disease. Moreover, such cancers would become rapidly refractory to chemotherapy, as witnessed by the increased tolerance of mismatch repair-deficient cells to alkylating agents (33), as well as to other DNA-modifying drugs such as cis-platin (34). Anecdotally, alkylating agents are often used together with MTX in chemotherapy cocktails, and it is possible that the two classes of drugs might act synergistically to select for mismatch repair-deficient and, therefore, drug-resistant clones.
These results, together with the data presented above, provide new evidence that mismatch repair deficiency can arise not only as the result of mutations or because of transcriptional down-regulation of mismatch repair genes (see ref. 35 for review), but that imbalance in expression of the individual components of the mismatch repair machinery also can result in a severe malfunction of the repair process.
Colorectal Cancer
Colon cancer is particularly associated with MSH3 dysfunction, as well as mutations in hMSH2 or hMLH128.
From 2013 and Park et al discussed how the MSH3 gene undergoes frequent somatic mutation in colorectal cancers (CRCs) with MMR deficiency29.
Deficient DNA MMR is found in approximately 15% of human colorectal cancers (CRCs) that display a distinct tumour phenotype.
Among CRCs examined as part of the Cancer Genome Atlas project, MSH3 mutations were detected in 40% of hypermutated tumors of which three quarters showed MSI [14].
MSH3 may contribute to resistance to chemotherapy:
While MSH3 appears to play a role in CRC development, its ability to regulate chemosensitivity remains unproven. Evidence indicates that the MSH2/MSH3 heterodimer, known as MutSb, is involved in the repair of toxic DNA double strand breaks (DSBs) induced by interstrand crosslink(ICLs) that are formed by agents such as cisplatin and psoralen[18]. The ability of homologous recombination (HR) to repair ICLs is dependent on MutSb, but not on MutSa or MLH1.Recent data suggest that MSH3 is involved in the repair of oxaliplatin-induced DNA ICLs [19,20,21], and co-localizes to DSB lesions that were induced by laser treatment [22] or by a carcinogen [18].
When expressed to excess then MMR can be inhibited, leading to carcinogenesis but if expressed in otherwise deficient cancer cells then your chemotherapy may fail. Balance is the key here:
Furthermore, MSH3-deficient cells were more susceptible to radiation-induced DNA DSBs, as shown by higher levels ofpH2AX, pChk2 and 53BP1 (marker of nonhomologous end joining), and apoptosis after oxaliplatin or SN-38 treatment. Accordingly, MSH3-deficient cells may become overwhelmed by the extent of DNA damage and undergo apoptosis whereas MSH3 proficiency provides protection from DSBs. In support of our data in isogenic HCT116 cells, we also generated SW480 colon cancer cells with suppression of MSH3. Similar results were found for the effect of MSH3 status upon chemosensitivity, suggesting that our results can be generalized.
DNA double-strand breaks (DSBs) induced by radiotherapy could also be less effective at inducing apoptosis due to excess MSH3:
HR: Homologous recombination.
RAD51: A DSB repair protein that also protects cells from radiation-induced cell death.
Tumor cells utilize their DNA repair capacity to prevent an accumulation of lethal DNA DSBs from cytotoxic chemotherapy or radiation [40]. Studies in yeast indicate that MSH3 can enable HR to repair DSBs [48]. After exposure to radiation, RAD51 rapidly forms a complex with BRCA2 and other proteins that stimulate RAD51-mediated strand exchange and the assembly of subnuclear foci characteristic of HR [49]. In addition to repair of radiation-induced DNA damage, RAD51 is involved in the repair of DNA DSBs produced by cisplatin, camptothecins and inhibitors of PARP [50,51,52]. In our study, the percentage of cells with >10 RAD51 foci was significantly decreased after radiation in MSH3-defieicnt [sic] vs-proficient cells, suggesting that MSH3 can regulate HR. In a recent study, MSH3-deficiency resulted in EMAST and pathway analysis revealed overexpression of proteins involved in DSB repair (MRE11 and RAD50) and apoptosis in HCT116 andHCT116+ch3 cells [53].
If your tumour type is deficient in MSH3 then consideration should be given to avoid therapeutics that may increase cellular concentrations, which would by implication preclude administration of synthetic mRNA gene agents which encode for this:
These data have clinical implications in that they may influence the selection of chemotherapeutic agents based upon tumor MSH3 status, and provide a rationale for the evaluation of MSH3 as a predictive biomarker in CRC patients.
In 2009, Haggar & Boushey discussed colorectal cancer epidemiology, including incidence, mortality, survival, and associated risk factors30.
Key takes:
Colorectal cancer is a major cause of morbidity and mortality throughout the world.1 It accounts for over 9% of all cancer incidence.2,3It is the third most common cancer worldwide and the fourth most common cause of death.2 It affects men and women almost equally, with just over 1 million new cases recorded in 2002, the most recent year for which international estimates are available.1,4,5,6 Countries with the highest incidence rates include Australia, New Zealand, Canada, the United States, and parts of Europe. The countries with the lowest risk include China, India, and parts of Africa and South America.3
Colorectal cancer survival is highly dependent upon stage of disease at diagnosis, and typically ranges from a 90% 5-year survival rate for cancers detected at the localized stage; 70% for regional; to 10% for people diagnosed for distant metastatic cancer.11,17 In general, the earlier the stage at diagnosis, the higher the chance of survival.
The likelihood of colorectal cancer diagnosis increases after the age of 40, increases progressively from age 40, rising sharply after age 50.2,17 More than 90% of colorectal cancer cases occur in people aged 50 or older.13,17
Neoplastic polyps of the colorectum, namely tubular and villous adenomas, are precursor lesions of colorectal cancer.8 The lifetime risk of developing a colorectal adenoma is nearly 19% in the U.S. population.15Nearly 95% of sporadic colorectal cancers develop from these adenomas.19
Approximately 5 to 10% of colorectal cancers are a consequence of recognized hereditary conditions.18 The most common inherited conditions are familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC), also called Lynch syndrome. Genes responsible for these forms of inherited colorectal cancer have been identified. HNPCC is associated with mutations in genes involved in the DNA repair pathway, namely the MLH1 and MSH2 genes, which are the responsible mutations in individuals with HNPCC.2,26
Dysfunction of MLH1 and MSH2 genes is associated with a lifetime risk of CRC which is as high as 80%. Long term impairment puts you at a particularly high risk of CRC and other cancers:
HNPCC may account for ~2 to 6% of colorectal cancers.2,13The lifetime risk of colorectal cancer in people with the recognized HNPCC-related mutations may be as high as 70 to 80%,27,28and the average age at diagnosis in their mid-40s.13 MLH1 and MSH2 mutations are also associated with an increased relative risk of several other cancers, including several extracolonic malignancies, namely cancer of the uterus, stomach, small bowel, pancreas, kidney, and ureter.2
Breast Cancer
In 2017, Restrepo et al published their findings from using software that they developed called “RNA2DNAlign” to systematically quantify the allele expression of somatic variants in breast cancer samples from The Cancer Genome Atlas (TCGA).
Their analysis also shows that MSH3 overexpression can be added to the long list of pro-cancer factors induced by experimental synthetic mRNA gene agents31:
CGC: Cancer Gene Census.
We document significant allele-preferential expression of functional variants in CGC genes and across the entire dataset. Notably, we find frequent allele-specific overexpression of variants in tumor-suppressor genes. We also report a list of over-expressed variants from non-CGC genes. Overall, our analysis presents an integrated set of features of somatic allele expression and points to the vast information content of the asymmetric alleles in the cancer transcriptome.
Next, we reviewed, on a per-gene basis, the current knowledge on the SOM-E genes and their possible implications in cancer. Despite not being listed in the CGC, some of these genes – such as MSH3 and NUAK1 and NFE2 – have been repeatedly linked to cancer31,32,33.
Another striking observation is that 6 of the genes with SOM-E variants –MSH3, RAD51, TCOF1, TP53BP1, CCNB2, and TOP3B – are directly implicated in DNA damage response and repair36,37,38,39 which was also the top-enriched pathway in the SOM-E dataset (p = 0.05, Metacore).
In summary, our research illustrates an important correlation between asymmetric alleles and cancer-implicated functionality…
Cancer Stem Cells (CSCs) and DNA Damage
MSH3 is associated with the resistance of CSCs to chemo and radiotherapy, and has implications for the likelihood of disease progression or recurrence.
Both embryonic and adult stem cells are endowed with a superior capacity to prevent the accumulation of genetic lesions, repair them, or avoid their propagation to daughter cells, which would be particularly detrimental to the whole organism. Inducible pluripotent stem cells also display a robust DNA damage response, but the stability of their genome is often conditioned by the mutational history of the cell population of origin, which constitutes an obstacle to clinical applications. Cancer stem cells are particularly tolerant to DNA damage and fail to undergo senescence or regulated cell death upon accumulation of genetic lesions. Such a resistance contributes to the genetic drift of evolving tumors as well as to their limited sensitivity to chemo- and radiotherapy.
…most neoplasms contain a population of cancer stem cells (CSCs) that drives oncogenesis and supports tumor progression (Kreso and Dick, 2014). Normal and malignant SCs share the ability to self-renew while generating differentiated cells (Blanpain and Simons, 2013).
Genetic lesions of endogenous or exogenous origin are major threats to the survival and function of SCs (Blanpain et al., 2011). SCs are particularly exposed to sources of DNA damage, and this can have catastrophic consequences for tissue and organismal homeostasis (Behrens et al., 2014). Thus, both normal and malignant SCs rely on a very robust DNA damage response (DDR), which—on the one hand—is beneficial as it preserves optimal SC function in healthy tissues, and—on the other hand—is detrimental as it favors the survival and resistance to treatment of CSCs (Mandal et al., 2011).
According to a debated model, CSCs are multipotent SCs responsible for the long-term clonal maintenance and growth of most human neoplasms (Kreso and Dick, 2014). CSCs—whose relative abundance is highly variable (2.5%–40%)—are normally identified based on CD24, CD44, CD117, or CD133 but display high degrees of genotypic, phenotypic, and functional heterogeneity. CSCs resemble ASCs as they can (co)exist in a cycling and quiescent state and as they acquire mutations that further increase heterogeneity (Kreso and Dick, 2014).
As compared to their relatively more differentiated counterparts, breast CSCs display reduced levels of oxidative DNA damage, both at baseline and after irradiation (Diehn et al., 2009).
MSH2-MSH3 Levels and Genomic Instability
A preprint by Medina-Rivera et al (2022) discussed how they used yeast cells and plasmids to demonstrate that elevated levels of the heterodimer MSH2-MSH3 were associated with genomic instability. They showed that DNA replication and base excision repair (BER) were interfered with in vivo in an expression and ATP-dependent manner.
This was consistent with their hypothesis that protein abundance and ATPase activities in MSH3 are the key drivers of MSH2-MSH3-mediatedgenomic instability.
They demonstrated that MSH2-MSH3 competes with both RAD27 and LIG1 for binding to DNA substrates, resulting in the inhibition of RAD27 endonuclease activity, ligation (the joining of two DNA strands or other molecules by a phosphate ester linkage) and a significant reduction of Okazaki fragment processingin vitro (short sequences of DNA nucleotides which are synthesized discontinuously and later linked together by the enzyme DNA ligase to create the lagging strand during DNA replication). Uncontrolled binding of MSH2-MSH3 to 5’ flap intermediates poses a potential risk for normal DNA metabolism.
They found that even low levels of MSH2-MSH3 overexpression can result in sensitivity to an alkylating drug called methyl methanesulfonate (MMS), resulting in lesions that could normally be repaired via BER.
And defects in cell cycle progression may be a result of Okazaki fragment stress.
The authors concluded that elevated levels of MSH2-MSH3 interfered with normal DNA metabolism, not just by binding DNA substrates but by engaging in aberrant MMR in an ATP binding-dependent manner. Their key take33:
…tight regulation of the Msh2-Msh3 expression levels is important in vivo to prevent interference with both DNA synthesis and the processing of a variety of DNA structures.
The increased tendency for DNA mutations (changes) and other genetic changes to occur during cell division. Genomic instability is caused by defects in certain processes that control the way cells divide. It occurs in many types of cancer. These defects may include mutations in certain genes involved in repairing damaged DNA or mistakes that don’t get corrected when DNA is copied in a cell. They may also include defects such as broken, missing, rearranged, or extra chromosomes. Studying genomic instability may help researchers understand how certain diseases, such as cancer, form. This may lead to new ways to diagnose, treat, and prevent disease.
A meta-analysis from 2015 by Miao et al associates polymorphism of MSH3 rs26279 with a significantly increased risk of cancer overall, especially for colorectal cancer and breast cancer34:
If MSH3 is indeed being expressed by both SARS-CoV-2 and synthetic mRNA gene agents, as the evidence presented so far strongly suggests, then it would appear that they chose and approved the perfect tool for increasing cancer incidence and neurological disorders over the coming years.
MSH3 and Neurological Disorders
In 2022, Bermingham et al filed a patent for “useful compositions and methods to treat nucleotide repeat expansion disorders35”, the focus being to therapeutically reduce MSH3 mRNA expression.
Huntington's disease: An inherited disorder that causes nerve cells (neurons) in parts of the brain to gradually break down and die. The disease attacks areas of the brain that help to control voluntary (intentional) movement, as well as other areas36.
Fragile X syndrome (FXS): Also known as Martin-Bell syndrome, FXS is an inherited condition that causes developmental delays, intellectual disabilities, learning and behavioral issues, physical abnormalities, anxiety, attention-deficit/hyperactivity disorder and/or autism spectrum disorder, among other problems37.
Key takes:
Nucleotide repeat expansion disorders (e.g., trinucleotide repeat expansion disorders) are generally categorized by the type of repeat expansion. For example, Type 1 disorders such as Huntington's disease are caused by CAG repeats which result in a series of glutamine residues known as a polyglutamine tract, Type 2 disorders are caused by heterogeneous expansions that are generally small in magnitude, and Type 3 disorders such as fragile X syndrome are characterized by large repeat expansions that are generally located outside of the protein coding region of the genes. Nucleotide repeat expansion disorders (e.g., trinucleotide repeat expansion disorders) are characterized by a wide variety of symptoms such as progressive degeneration of nerve cells that is common in the Type 1 disorders.
The present inventors have found that inhibition or depletion of MSH3 level and/or activity in a cell is effective in the treatment of a nucleotide repeat expansion disorder (e.g., a trinucleotide repeat expansion disorder). Accordingly, useful compositions and methods to treat nucleotide repeat expansion disorders (e.g., a trinucleotide repeat expansion disorder)…
MSH3, another component of the mismatch repair pathway, has been reported to be linked to somatic expansion: polymorphisms in Msh3 was associated with somatic instability of the expanded CTG trinucleotide repeat in myotonic dystrophy type 1 (DM1) patients (Morales et al., (2016) DNA Repair 40: 57-66).
There is also evidence that MSH3 mRNA can be distributed systemically in cerebrospinal fluid (CSF) derived exosomes38:
Irina Antonijevic, MD, PhD & Peter Bialek, PhD, Triplet Therapeutics, Inc.
Triplet is harnessing the growing evidence that DNA damage response (DDR) genes are potent modifiers of onset and rate of progression of repeat expansion diseases (REDs), such as Huntington’s disease (HD), myotonic dystrophy, and spinocerebellar ataxias. DDR modifier genes act by modulating the rate of somatic expansion at disease-causing gene loci, such as mutant huntingtin (mHTT). The DDR gene MSH3 has been strongly associated with disease progression in HD, including in particular cognitive deterioration that begins in premanifest HD. Only about 50% lowering of MSH3 appears sufficient to halt or markedly slow somatic expansion of mHTT. As no untoward safety signals have been associated with this moderate level of MSH3 lowering, MSH3 has emerged as a prime target with the potential to treat HD and multiple other REDs by acting upstream of individual disease genes.
They assayed MSH3 mRNA to show a comparable dose-dependent reduction in MSH3 mRNA in cells and secreted exosomes:
In vitro PoC with TTX-3360 was achieved, showing a comparable dose-dependent reduction in MSH3 mRNA in cells and secreted exosomes. Further, MSH3 mRNA was detected in CSF exosomes at similar levels in healthy and premanifest HD subjects. Ongoing analyses of longitudinal studies in NHPs with repeat CSF sampling will inform on the correlation between MSH3 mRNA in CSF exosomes and brain tissues after treatment with TTX-3360. The initial 48-week follow-up data from SHIELD HD and progress on the target engagement biomarker assay will be presented.
And finally, in 2022 Sfera et al (incl. Sabine Hazan md) published “The MSH3 Gene From A Neuropsychiatrist’s Perspective39” in response to reading “MSH3 Homology and Potential Recombination Link to SARS-CoV-2 Furin Cleavage Site”.
This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Key takes:
Aside from its well-established role in averting tumor genesis, novel preclinical studies found that MSH3 is a key regulator of short tandem repeats (STRs), DNA sequences characteristic of monogenic neuropsychiatric disorders, such as Huntington’s disease (HD) and fragile X syndrome (FXS) [2-4]. For example, FXS is caused by CGG repeats in the fragile X messenger ribonucleoprotein 1 (FMR1) gene [5]. Interestingly, the designers of COVID-19 mRNA vaccine chose to encode the 42 arginine residues (found in viral S protein) via a rare CGG codon, increasing the odds of STRs formation [6]. Many viruses, including SARS-CoV-2, human cytomegalovirus (HCV), and human immunodeficiency virus (HIV), were demonstrated to generate STRs, increasing the risk of tandem repeat disorders (TRDs) [7-12]. On the other hand, fragile X mental retardation protein (FMRP), the product of FMR1 gene, was demonstrated to target influenza and Zika viruses, while metabotropic glutamate 5 receptor (mGluR5) inhibitors, commonly utilized in FXS, often ameliorate SARS-CoV-2 symptoms, indicating a two-way street between viral infections and TRDs [13-15].
Short tandem repeats are associated with an increased risk of schizophrenia and autism spectrum disorder:
Recent studies have reported that STRs can increase the risk of schizophrenia and autism spectrum disorder (ASD), indicating that these sequences play a major role not only in monogenic but also in polygenic disorders [16,17]. Indeed, the findings of Ambati BK, et al. are in line with our own studies that connected FCS to pathological cell-cell fusion, neurodegeneration, and psychopathology [18,19].
Pfizer and Moderna COVID-19 messenger RNA (mRNA) vaccines are heavily engineered to facilitate translation and improve stability, modifications that include codon optimization enriched with CG repeats [6]. However, as MSH3 regulates STRs, including the CG repetitions, vaccine efficacy is likely enhanced by the inhibition or attenuation of this protein. This may explain the reason Moderna was interested in patenting this molecule in 2016. In addition, as COVID-19 mRNA therapeutics encode the entire S antigen, MSH3 may be over expressed, a phenomenon associated with loss of function [23]. Indeed, MSH3 may be inactivated via promoter methylation or over expression [24].
From the neuropsychiatric perspective, the novel MSH3 findings are significant as this protein, encoded on chromosome 5 (q11-q13), shares a common promoter with dihydrofolate reductase (DHFR), a gene disrupted in many neuropsychiatric conditions,including ASDs, schizophrenia, depressive and bipolar disorder as well as immune dysfunction, diabetes, type I, and epilepsy [25-31]. Due to the common promoter, vaccine-induced MSH3 inhibition likely attenuates DHFR, predisposing to these pathologies. In favor of this statement, we bring the fact that treatment with methotrexate, a DHFR inhibitor, was associated with neuropsychiatric pathology, including anxiety, depression, suicidal behavior, and dementia [16], [32-36].
Therapeutics
Quick take is that prevention is better than cure, but its never too late to start.
In 2023, De et al published a very useful meta-analysis of natural sources of drugs with anti-colorectal cancer (CRC) properties40.
Key takes:
Chemoprevention aims to intervene, prevent, suppress, and reverse the initiation and progression of carcinogenesis. It further attempts to decrease the recurrence of cancer through the usage of drugs, vitamins, and nutritional supplements [66]. Various agents, including nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin, and other agents, such as metformin, statins, minerals, and vitamins, have been previously studied for their chemopreventive benefits regarding CRC (Table 2). There is little doubt that a significant stride has been made into the unventured territories for the chemoprevention of CRC.
In CRC involving the APC/β-catenin pathway, cyclooxygenase-2 (COX-2) is often implicated in the early and later stages of the adenoma sequence, driving the formation into a carcinoma [120,121,122,123]. Furthermore, COX-2 overexpression produces vascular endothelial growth factor (VEGF), which promotes tumor angiogenesis [124,125]. Hence, by targeting COX-2, various studies have shown that NSAIDs, ranging from aspirin and sulindac to the more selective COX-2 inhibitors, such as celecoxib, have proven benefits in reducing disease risk [126,127]. In the 1990s, the U.S. Preventive Services Task Force recommended aspirin to prevent non-high-risk CRC [128,129,130].
Other drugs, such as metformin, showed promising effects in reducing the risk of CRC development. Recent meta-analyses showed that metformin could reduce CRC risk by 22% [131]. In an ongoing ASAMET trial for the tertiary prevention of stage I–III CRC, patients were administered low doses of aspirin combined with metformin for a potential synergistic chemo-preventive action [132].
As discussed here, the phytochemicals were reported to act through inhibiting hallmarks of various CRC attributes, such as the potential of cell growth and proliferation, self-renewal, invasion, migration, and angiogenesis through inducing apoptosis, ferroptosis, and autophagy-mediated cell death pathways (Figure 6). These activities involved the modulation of various pathways, such as the levels of proinflammatory cytokines and chemokines (IL-1, IL-6, ICAM-I, TNF, COX-2, iNOS, KC, and MCP1), oxidative stress markers and pathways (SOD, catalase, thiolase, glutathione peroxidase, GSH and Keap1/NRF2/GSK-3β/HO-1), cell cycle regulators (cyclin D1, cyclin E, and CDK 4/6), apoptotic/autophagy regulators (p21, p53, caspase-3, caspase-9, Bax, Bcl-2, Bak, and Beclin1), proliferative signaling pathways regulators (PI3K/Akt/mTOR/AMPK, Wnt/β-catenin, MAPK-p38, ERK, MEK, and c-Myc), regulators of invasion, migration, metastasis, and angiogenesis (Notch1, STAT-3, VEGF, CD31, MMP-2, MMP-3, MMP-9, MMP-16, EGFR, Twist1, Vimentin, FMS-related tyrosine kinase 4, endothelial growth receptor-3, Snail, N-cadherin, E-cadherin, TIMP-1, and TIMP-2), stemness (CD133, CD44, ALDH1, CD29, DCLK-1, and LGR5) and expression of tumor suppressive miRNAs (miR34a, miR200c, and miR145).
Baicalein suppressed AOM/DSS-induced colon tumors in mice and induced apoptotic cell death. Baicalein suppressed inflammation by PARP1-mediated NF-κB inhibition [180]. A dose of 50 mg/kg baicalin suppressed the growth of highly metastatic SW620 tumor xenograft in BALB/c nude mice [181]. Baicalin inhibited the TLR4/NF-κB signaling and significantly suppressed CT-26 tumor growth, migration, and invasion. Anti-tumor immunity was also enhanced by an increase in CD4+ and CD8+ T cells in CT-26 tumors [182]. Baicalein treatment induced apoptosis in a p53-mediated Akt-dependent manner and suppressed HT-29 tumor xenograft [183]. In another study, baicalein suppressed MMP-2 and MMP-9 and inhibited DLD1 tumor growth and metastatic effects by inhibiting phosphorylation of ERK [184].
Caffeic acid (3,4-dihydroxycinnamic acid) is a nonflavonoid catechol with potent antioxidant properties. It is found in almost all plants as an intermediate in the lignin biosynthesis pathway. The prime source of caffeic acid is coffee. Caffeic acid possesses various pharmacological properties, such as antioxidant, anti-inflammatory, anticancer, and neuroprotective effects [400]. Caffeic acid, by direct interaction, inhibited MEK1 and TOPK activity in an ATP non-competitive manner. Kang et al. [303] conducted experiments using caffeic acid on a mouse tumor model. It demonstrated action by inhibition of ERK and p90RSK activation. Caffeic acid suppressed the TPA-induced activation of AP1, NF-κB, and ERK signaling, and thus neoplastic transformation induced by TPA, EGF, and H-Ras. Through inhibition of ERK functions, caffeic acid inhibited lung metastasis of CT-26 cells. This study also indicated the usefulness of caffeic acid in reducing ERK activity in patient tumor samples.
Caffeic acid effectively inhibited cancer stem cells (CSC) and reduced radiation-induced sphere formation of CD133+ and CD44+ CSC in two patient-derived tumor xenograft (PDTX) models of human CRC in immune-suppressed mice. In vivo, the radiation-induced elevation of PI3K/Akt signaling pathway was also suppressed by caffeic acid. In caffeic acid-treated xenograft samples, the abundance of CD133+ and CD44+ subpopulations of CSC cells were decreased. In addition, CD44+ and CD133+ cells of CRC lost their ability for self-renewal, migration, and CSC-like properties due to caffeic acid in a PDTX xenograft model. Inhibition of PI3K/Akt signaling was described as a significant mode of action caffeic acid in inhibiting CSC proliferation [304].
CAPE and caffeic acid p-nitro-phenethyl ester (CAPE-pNO2) upregulated the levels of p53, p27, p21, cytochrome c (cyt. C), and cleaved caspase-3, but downregulated procaspase-3, Cdk2, and c-Myc in HT-29 tumor xenograft in mice. There was a dose-dependent inhibition of tumor growth and VEGF expression by these compounds, with no visible toxicity to normal cells [306].
Consumption of decyl caffeic acid inhibited tumor growth in mice with a HCT116-induced tumor xenograft. The mechanism of action involved the induction of cell cycle arrest at the S phase as well as autophagy [307].
The downregulation of COX-2 levels can be achieved upon treatment with EGCG [206], curcumin [194,197], kaempferol [239], luteolin [242,243], myricetin [259], naringenin [262], piceatannol [342], pterostilbene [344], syringic acid [326], boeravinone B [191], hesperidin [227], isoliquiritigenin [235], orientin [268], quercetin [281], and xanthohumol [301]. Caffeic acid suppressed TPA-induced activation of AP1 and NF-κB signaling [303]. Many phytophenols can induce an antioxidant response, such as EGCG, gallic acid, boeravinone B, eriodyctyol, luteolin, and morin. Caffeic acid phenethyl ester and caffeic acid phenylpropyl ester-induced mTOR inhibition through the activation of AMPK [305]. Isoangustone A upregulated AMPK phosphorylation in vivo [234]. Pterostilbene inhibited EGFR in an AOM-induced colonic adenomas in mice [344].
Current treatment strategies
Genetic and molecular basis of colorectal cancer along with the current treatment strategies where potentials of phenolic compounds were indicated. CIN, MSI and CIMP are the prime factors in CRC development. Besides the currently available chemotherapeutic treatment strategies, different polyphenols are reported to induce CRC cell death by apoptosis and/or autophagy and/or necrosis. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9913554/
There is increasing evidence in favor of the idea that diet can influence the intestinal microbiome and thus the risk of CRC. Diets rich in fruits and vegetables can be associated with gut microbiome rich in Prevotella compared with Bacteroides associated with good colonic health while the consumption of diet with low plant-based food rich in processed food led to the opposite effects [411,412]. Diets rich in plant-based nutraceuticals could regulate host immune and inflammatory behavior and thus gut homeostasis through modulating the composition and functionality of the gut microbiome [413]. Therefore, CRC incidence and progression can be reduced by modulating gut microbiome by careful choice of diet and phytochemicals which could be a promising and efficient way to reduce the burden of CRC [413]. Gut microbiota can digest dietary phytochemicals by their unique ability to produce short chain fatty acids, such as butyric acid, with anti-inflammatory and antineoplastic activity [414]. Phenolic phytochemicals have served us as an important source of novel drugs/leads.
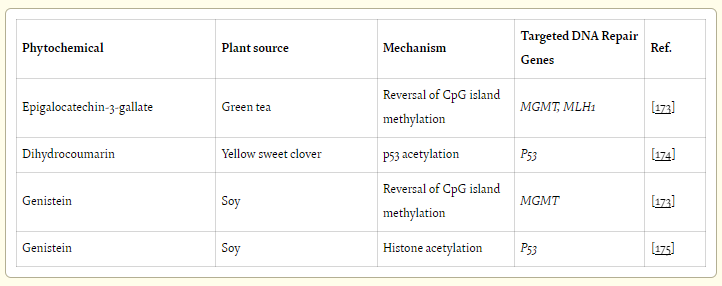
Phytochemicals involved with DNA damage repair were reviewed by Bernstein, Prasad & Bernstein (2012)41:
Some polyphenols affect expression of many genes, including DNA repair genes, through epigenetic alterations, as reviewed by Link et al. [172]. Examples of DNA repair genes expression increased by epigenetic alteration are listed in Table 9.
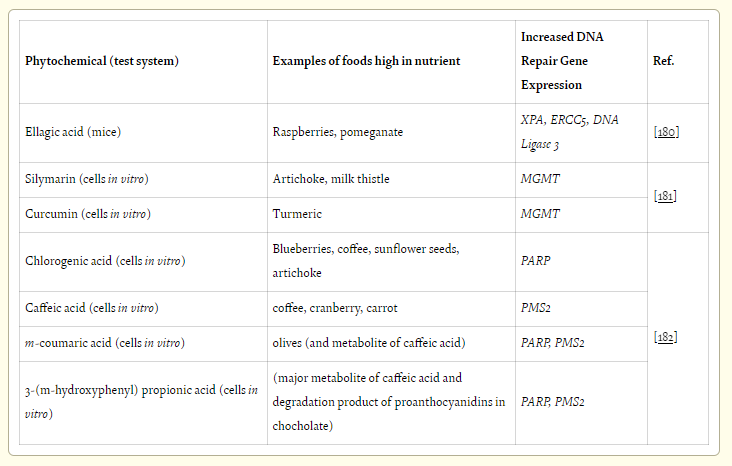
A recent review article by Collins et al. [176] summarizes some examples of micronutrients that affect DNA repair gene expression, though by unknown mechanisms. Table 10 lists such phytochemicals, without defined mechanisms, that increase DNA repair gene expression, along with commonly known foods that are high in those phytochemicals [177, 178, 179].
Bernstein et al. [182] evaluated antioxidants based on their ability to increase DNA repair proteins PARP-1 and Pms2 in vitro. They tested 19 anti-oxidant compounds and of these 19 compounds only chlorogenic acid and its metabolic products: chlorogenic acid, caffeic acid, m-coumaric acid and 3-(m-hydroxyphenyl) propionic acid, increased expression of the two tested DNA repair genes in HCT-116 cells (Table 10).
DCA: Deoxycholic acid.
CGA: Chlorogenic acid.
When DCA is added to the diet of wild-type mice to raise the level of DCA in the mouse feces to the level in feces of humans on a high fat diet, by 10 months of feeding 94% of the mice develop tumors in their colons with 56% developing colonic adenocarcinomas [60]. This mouse model develops tumors solely in the colon, phenotypically similar to development of colon cancer in humans. When CGA, equivalent to 3 cups of coffee a day for humans, was added to the DCA supplemented diet it was dramatically protective against development of colon cancer, reducing incidence of colon cancer significantly from 56% to 18% [60].
Curcumin
And from 2004, van Erk et al conducted an in vitro investigation into the effects of curcumin exposure on two human cell lines42.
Key takes:
Early response genes were defined as genes that were differentially expressed after exposure to curcumin for 3 or 6 hours. A selection of the early response genes in HT29 cells is listed in table table11 (low concentration) and table table22 (high concentration). Early response genes were involved in several processes, including cell cycle control, signal transduction, DNA repair, transcription regulation, cell adhesion and xenobiotic metabolism. Both curcumin concentrations caused an increase in expression of genes involved in DNA repair, e.g. MLH1, MSH3 and ERCC2 (Tables (Tables11 and and2).2). Upregulated signal transduction genes included STAT3 and STAT5b (Table (Table1)1) and some genes of the MAPK signal transduction pathway (MAP3K10, MAP4K2; table table11 and and2).2). Some other MAPK signal transduction genes were downregulated by the low curcumin concentration (Table (Table1).1). Expression of a group of genes involved in cell adhesion and protein binding was induced by short-term exposure to curcumin, including annexin (Table (Table1)1) and integrin genes (Table (Table2).2). Several genes involved in xenobiotic metabolism were downregulated after short-term exposure to the high concentration of curcumin, namely GSTT2, GSTM4, CYP1B1 (Table (Table2).2). Expression of GCLC, involved in glutathione synthesis, was upregulated after 6 hours (Table (Table22).
Also, genes involved in transcription regulation were induced or repressed by short-term exposure to curcumin. Among these were transcription factors such as activating transcription factor 4 (ATF4) and early growth response 1 (EGR1) (Table (Table2).2). One of the target genes regulated by ATF4 is asparagine synthetase (ASNS) [31]. Figure22 shows the expression profile of ATF4 and ASNS in response to curcumin. Induction of ATF4 expression at the early time points is followed by induction of expression of ASNS at the same and later time points. EGR1, a transcription factor involved in cell growth regulation and tumor suppression [32], was the most upregulated early response gene. In contrast to this strong upregulation after exposure to the high concentration of curcumin, this gene was not induced by the low curcumin concentration (Figure (Figure3A).3A).
When looking in detail into the gene expression profiles at the different time-points, several genes known to be involved in colon carcinogenesis were found that responded to curcumin exposure. Expression changes of these genes are shown in figure figure3.3.
EGR1, a transcription factor involved in cell growth regulation and tumor suppression [32], was the most upregulated early response gene. In contrast to this strong upregulation after exposure to the high concentration of curcumin, this gene was not induced by the low curcumin concentration (Figure (Figure3A).3A).
Expression profile of genes in response to curcumin; grey bars indicated the low concentration and black bars indicate the high concentration. EGR1: early growth response 1; PLK: polo-like kinase; TP53: tumor protein p53; CA2: carbonic anhydrase 2; MTHFD2: methylene tetrahydrofolate dehydrogenase/cyclohydrolase; AKT1: protein kinase B/Akt; PLAU: urokinase-type plasminogen activator; PLAUR: urokinase-type plasminogen activator receptor; TM4SF1: transmembrane 4 superfamily member 1; TM4SF4: transmembrane 4 superfamily member 4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC421747/
In addition to the functional groups mentioned above several genes involved in the cell cycle or cell growth were among the early response genes. For example several growth factors (AREG, VEGF, FGFR1) were upregulated three hours after exposure to the high concentration of curcumin (Table (Table2).2).
One of the most strongly downregulated genes in HT29 cells was polo-like kinase (PLK), after exposure to the high curcumin concentration for 24 hours (fig. (fig.3B).3B). PLK is a cell cycle gene involved in spindle assembly. It is expressed at a higher level in colorectal cancer than in normal colon tissue [33]. Downregulation of PLK has been shown to inhibit cell growth in cancer cells [34]. Several histone genes (H3F3A, HIST1H4C) were downregulated by curcumin, especially at the 12 h and 24 h time points (data not shown).
In contrast to cell cycle-related genes, only a few genes involved in apoptosis were differentially expressed in response to curcumin. The most striking effect was observed with programmed cell death 2 (PDCD2), which was downregulated 1.8-fold by the high concentration of curcumin after exposure for 12 hours.
Curcumin is an anti-inflammatory agent and can act as a natural non-steroidal anti-inflammatory drug (NSAID). Some of the genes differentially expressed in response to curcumin in our study were also differentially expressed in human colon cancer cells lines in response to other non-steroidal compounds with anti-inflammatory action (NSAIDs) such as aspirin or sulindac [76].
In invasive human colonocytes butyrate inhibited urokinase plasminogen activator (uPA) activity, and downregulated PCNA and TP53 levels after exposure for 12–18 hours [77]. In our study a similar response was found, as curcumin reduced expression of PCNA, TP53 and PLAU (uPA). This indicates that there may be some overlap in response and in mechanism of action between different NSAIDs like curcumin, aspirin and sulindac, but also between NSAIDs and butyrate. However, both aspirin and sulindac increased expression of several metallothionein genes, which were downregulated by curcumin in our study [76].
Some known effects of curcumin were confirmed (G2/M cell cycle arrest, induction of phase-II genes) and the existing knowledge was extended with extra information (e.g. time points of the observed changes and genes involved or linked to these physiological effects). Also, potential new leads to mechanisms explaining the biological activity of curcumin were identified, for example the effect on tubulin genes and differential expression of p16(INK4)/TP53/RB1. Studying expression changes of thousands of genes has provided increased insight into the mechanism of action of curcumin in colon cancer cells, helping us to understand how this compound can protect against development of colon cancer.
Baicalin
In 2016 Zhang et al screened 289 compounds that act by inducing DNA replication stress or DNA damage and shortlisted the flavone baicalein.
A link to a literature review of baicalin is in the above Substack and cited below43.
Key takes from “A novel chemotherapeutic agent to treat tumors with DNA mismatch repair deficiencies”44:
Impairing the division of cancer cells with genotoxic small molecules has been a primary goal to develop chemotherapeutic agents. However, DNA mismatch repair (MMR)-deficient cancer cells, are resistant to most conventional chemotherapeutic agents. Here we have identified baicalein as a small molecule that selectively kills MutSα-deficient cancer cells. Baicalein binds preferentially to mismatched DNA and induces a DNA damage response in a mismatch repair-dependent manner. In MutSα-proficient cells, baicalein binds to MutSα to dissociate CHK2 from MutSα leading to S phase arrest and cell survival. In contrast, continued replication in the presence of baicalein in MutSα-deficient cells results in a high number of DNA double-strand breaks and ultimately leads to apoptosis. Consistently, baicalein specifically shrinks MutSα-deficient xenograft tumors and inhibits the growth of AOM-DSS-induced colon tumors in colon-specific MSH2 knockout mice. Collectively, baicalein offers the potential of an improved treatment option for patients with tumors with a DNA MMR deficiency.
Hypothesizing that these compounds could provide a better treatment option for Lynch Syndrome tumors, we tested the ability of the compounds to selectively kill cells with mutations in the MSH2 gene. From this screen, we identified baicalein as a small molecule capable of inducing apoptosis in MSH2-deficient cells both in vitro and in vivo. Baicalein is a flavone derived from the roots of Scutellaria baicalensis and Scutellaria lateriflora. Previously, baicalein was used in some Asian counties as herbal supplements to enhance liver health. Recent years, more and more studies of baicalein have proceeded on many human diseases related areas, such as cancer and diabetes(16–20). Although baicalein has previously been reported to have anti-tumor activities(18,19,21), this manuscript identifies a novel mechanism of action for baicalein in which the compound acts on the MutSα/CHK2/ATM pathway as well as preferentially binding to the mismatched DNA.
As we have all been exposed to synthetic or viral mRNA at some point then MSH3 overexpression has and likely will continue to be a risk for us all. If you are relatively young, without co-morbidities and a normal, functioning immune system then hopefully any aberrant cells with unrepaired DNA damage will be identified and eliminated by other parts of an active immune system (eg CD4+ & CD8+ T-cells), before they can develop into a symptomatic disease state.
But to apply the precautionary principle it would be advisable to try to follow as close to a healthy lifestyle as you can as early as possible, whilst incorporating therapeutics with antiviral and anticancer properties into your diet to further mitigate any risk.
I would also recommend extreme caution before being administered any new fast-tracked allopathic drugs, gene therapies or synthetic foods as long term clinical trials are no longer the norm and what trials data there is cannot be relied upon due to regulatory capture, use of synthetic data45 and the withholding of unsupportive data.
DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. Without your support I would be unable maintain this Substack. To receive new posts and support my work, consider becoming a free or paid subscriber.
(lightly redacted where he mentioned an individual)
DC your post is a massive piece of work so well done, and I admit to only skimming it BUT saw a couple of issues if you don't mind.
(1) it was me that put the "BLAST" article out in the public domain in Dec 2021 that allowed the Ambati paper to be published (it was previously being suppressed... but not mentioned in the FCS section 😥… …
(2) The GENOME sequence of the FCS in the vaccine is different from the one in the virus and therefore is NOT in the Moderna patent. The resulting amino acid sequence however is maintained through codon optimisation. Surprisingly, although codon optimisation is meant to increase GC content - they REDUCED it in the FCS portion. Presumably to avoid any comparisons to the moderna sequence (and assuming the vaccine was produced in advance, many months prior)
(3) Although the MSH3 moderna patent is for a mutated MSH3, the bit that transferred into the virus and vaccine is only one epitope. I can't remember now if the same epitope exists in human MSH3 (non mutated). Either way, transfecting with this spike gene is not likely to give you a mutated MSH3 in your somatic cells. We don't know what mechanisms of action the spike protein have on DNA repair outside of the Jiang-Mei paper, but that was shown to impact the p53 HRD pathway (not the MMR pathway). In other words, it's a stretch to claim that the inclusion of the FCS from a Moderna patent should have any impact on your MMR pathway. By the way the MMR pathway genes we look for in bowel and uterine cancers are MLH1, MSH2, MSH6 and PMS2.
My response:
We need to see more experimental and especially clinical evidence to find out exactly what is being expressed from whatever source, whether codon optimised versions or not, as attenuation may have been the objective?
Returning to the Sfera et al paper from 2022:
Pfizer and Moderna COVID-19 messenger RNA (mRNA) vaccines are heavily engineered to facilitate translation and improve stability, modifications that include codon optimization enriched with CG repeats [6]. However, as MSH3 regulates STRs, including the CG repetitions, vaccine efficacy is likely enhanced by the inhibition or attenuation of this protein. This may explain the reason Moderna was interested in patenting this molecule in 2016. In addition, as COVID-19 mRNA therapeutics encode the entire S antigen, MSH3 may be over expressed, a phenomenon associated with loss of function [23]. Indeed, MSH3 may be inactivated via promoter methylation or over expression [24].
Even if not overexpressing full MSH3 the generation of autoantibodies to the related sequences that are in there may put us at risk of a range of Lupus like disorders, and any consequential imbalance in MSH3 can also affect other DNA MMR proteins, as discussed previously.
From “DNA mismatch repair enzymes: Genetic defects and autoimmunity” by Muro et al (2015)46:
DNA mismatch repair (MMR) is one of the several DNA repair pathways conserved from bacteria to humans. The primary function of MMR is to eliminate the mismatch of base–base insertions and deletions that appear as a consequence of DNA polymerase errors at DNA synthesis. The genes encoding the DNA MMR enzymes (MMREs) are highly conserved throughout evolution. In humans, there are two sets of MMREs, corresponding to homologues of the bacterial MutLS systems. The human MutS enzymes consist of MSH2, MSH3 and MSH6, and the human MutL enzymes include MLH1, MLH3, PMS1 and PMS2. Since the beginning of this century, a few reports on autoantibodies to some MMREs have been reported in autoimmune inflammatory myopathy, cancer and hematological disorders.
Disclaimer
This site is strictly an information website about potential therapeutic agents and a review of associated research papers. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
Sattar S, Kabat J, Jerome K, Feldmann F, Bailey K, Mehedi M. Nuclear translocation of spike mRNA and protein is a novel pathogenic feature of SARS-CoV-2.bioRxiv. Published online September 27, 2022:2022.09.27.509633. doi:10.1101/2022.09.27.509633
Frolova EI, Palchevska O, Lukash T, Dominguez F, Britt W, Frolov I. Acquisition of Furin Cleavage Site and Further SARS-CoV-2 Evolution Change the Mechanisms of Viral Entry, Infection Spread, and Cell Signaling.J Virol. 96(15):e00753-22. doi:10.1128/jvi.00753-22
Mekawy AS, Alaswad Z, Ibrahim AA, Mohamed AA, AlOkda A, Elserafy M. The consequences of viral infection on host DNA damage response: a focus on SARS-CoVs. J Genet Eng Biotechnol. 2022 Jul 13;20(1):104. doi: 10.1186/s43141-022-00388-3. PMID: 35829826; PMCID: PMC9277982.
Örd M, Faustova I, Loog M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV.Sci Rep. 2020;10:16944. doi:10.1038/s41598-020-74101-0
Rhea EM, Logsdon AF, Hansen KM, et al. The S1 protein of SARS-CoV-2 crosses the blood–brain barrier in mice.Nat Neurosci. 2021;24(3):368-378. doi:10.1038/s41593-020-00771-8
Anandamide. Sequencing the Pfizer monovalent mRNA vaccines also reveals dual copy 72-bp SV40 Promoter. Nepetalactone Newsletter. Published April 12, 2023. Accessed July 14, 2023.
Nance KD, Meier JL. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines.ACS Cent Sci. 2021;7(5):748-756. doi:10.1021/acscentsci.1c00197
Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA.Immunity. 2005;23(2):165-175. doi:10.1016/j.immuni.2005.06.008
Karikó K, Muramatsu H, Welsh FA, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833-1840. doi:10.1038/mt.2008.200
Parr CJC, Wada S, Kotake K, et al. N 1-Methylpseudouridine substitution enhances the performance of synthetic mRNA switches in cells.Nucleic Acids Res. 2020;48(6):e35. doi:10.1093/nar/gkaa070
Kariko K, Weissman D. RNA containing modified nucleosides and methods of use thereof. Published online October 2, 2012. Accessed July 14, 2023. https://patents.google.com/patent/US8278036B2/en
Turner JS, O’Halloran JA, Kalaidina E, et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses.Nature. 2021;596(7870):109-113. doi:10.1038/s41586-021-03738-2
Craddock V, Mahajan A, Spikes L, et al. Persistent circulation of soluble and extracellular vesicle-linked Spike protein in individuals with postacute sequelae of COVID-19.J Med Virol. 2023;95(2):e28568. doi:10.1002/jmv.28568
Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene.Proc Natl Acad Sci U S A. 1998;95(15):8568-8573. doi:10.1073/pnas.95.15.8568
Park JM, Huang S, Tougeron D, Sinicrope FA. MSH3 Mismatch Repair Protein Regulates Sensitivity to Cytotoxic Drugs and a Histone Deacetylase Inhibitor in Human Colon Carcinoma Cells.PLOS ONE. 2013;8(5):e65369. doi:10.1371/journal.pone.0065369
Restrepo P, Movassagh M, Alomran N, et al. Overexpressed somatic alleles are enriched in functional elements in Breast Cancer.Sci Rep. 2017;7(1):8287. doi:10.1038/s41598-017-08416-w
Medina-Rivera M, Sridharan M, Becker J, et al. Msh2-Msh3 interferes with DNA metabolism in vivo. Published online September 6, 2022:2022.09.06.506750. doi:10.1101/2022.09.06.506750
Miao HK, Chen LP, Cai DP, Kong WJ, Xiao L, Lin J. MSH3 rs26279 polymorphism increases cancer risk: a meta-analysis.Int J Clin Exp Pathol. 2015;8(9):11060-11067.
US Patent Application for OLIGONUCLEOTIDES FOR THE TREATMENT OF NUCLEOTIDE REPEAT EXPANSION DISORDERS ASSOCIATED WITH MSH3 ACTIVITY Patent Application (Application #20230068672 issued March 2, 2023) - Justia Patents Search. Accessed July 14, 2023. https://patents.justia.com/patent/20230068672#description
De S, Paul S, Manna A, et al. Phenolic Phytochemicals for Prevention and Treatment of Colorectal Cancer: A Critical Evaluation of In Vivo Studies.Cancers (Basel). 2023;15(3):993. doi:10.3390/cancers15030993
Bernstein C, Prasad AR, Nfonsam V, et al. DNA Damage, DNA Repair and Cancer. In: New Research Directions in DNA Repair. IntechOpen; 2013. doi:10.5772/53919
van Erk MJ, Teuling E, Staal YC, et al. Time- and dose-dependent effects of curcumin on gene expression in human colon cancer cells.J Carcinog. 2004;3:8. doi:10.1186/1477-3163-3-8
Zhang Y, Fox JT, Park YU, et al. A novel chemotherapeutic agent to treat tumors with DNA mismatch repair deficiencies.Cancer Res. 2016;76(14):4183-4191. doi:10.1158/0008-5472.CAN-15-2974
Muro Y, Sugiura K, Mimori T, Akiyama M. DNA mismatch repair enzymes: Genetic defects and autoimmunity.Clinica Chimica Acta. 2015;442:102-109. doi:10.1016/j.cca.2015.01.014
Thank you so much for this ... Fantastic ... Very relevant to a few things I've been looking at... Thank you for putting it in easy to read format for those of us who only did basic microbiology (many years ago!)... Highly appreciated!






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
Thank you so much for this ... Fantastic ... Very relevant to a few things I've been looking at... Thank you for putting it in easy to read format for those of us who only did basic microbiology (many years ago!)... Highly appreciated!
Well, I guess I know how my next two days will be spent. Thank you.