


Megathread #32: SARS-CoV-2, spike protein, BRD4 and associated pathologies
or "If you don't like this cancer pathway I have others"


Updates
21st July ‘23: Links to biodistribution studies.
26th July ‘23: GP120 induced BRD4 expression.
BRD4 induced telomere lengthening of cancer cells.
Reading time: about 1 hour.
Also available in other languages:
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.

Contents
Experimental evidence for spike protein induced enhanced BRD4 expression
BRD4, neurological disorders and cardiovascular pathologies
Introduction
Its been a long time since I needed to post twice in one month. This review could almost be called “Megathread #31b” as its a continuation from the last Substack concerning pathologies and therapeutics related to spike protein inhibition of DNA mismatch repair mechanisms (MMR) via disruption to the MSH family:
But whereas with MSH3 we still need more experimental and clinical data for confirmation, with BRD4 the science is more settled and is well enough defined to stand on its own as a mediator of deleterious pathophysiological mechanisms.
To avoid repetition and to keep this review as short as possible please refer to the last Substack for background on protein synthesis, DNA mutations and therapeutics.
Most of us are by now familiar with the papers on spike protein translocating to the nucleus and impairing the key tumour suppressors BRCA1 & 2 and p53 but what about BRD4? There were many papers published in 2020-22 about blocking it to impede viral invasion, for example:

And in the last week I learned that BRD4 interacts with the host DNA damage repair mechanisms (DDR) too, and not in a good way either.
I’m always super interested in receptors to spike protein in particular, as these pathologies are more likely to explain and correlate with AE’s induced by mass administration of synthetic mRNA agents. It turns out that not only is BRD4 expression promoted by spike protein, but also by the E protein from COVID-19 viral infections:

Having multiple signalling pathways upregulating an oncogenic protein is never good, especially if boosters as well as breakthrough infections are contributory?
From the review “The consequences of viral infection on host DNA damage response: a focus on SARS-CoVs” by Mekawy et al (2022)1:

BRD2/4 interactions with SARS-CoV-2 E and spike proteins:
DSB: double-strand break repair.
Several bromodomain proteins (BRDs) get recruited for DSB repair through chromatin-remodeling complexes. For example, BRD2 binds to the acetylated histone 4 at the DSB site to protect it from histone deacetylases. BRD2 then allows for the recruitment of a second bromodomain protein, ZMYND8, which promotes the acetylation process [49]. Additionally, BRD4 recruits condensin II to remodel the acetylated histones, thus inhibiting DDR signaling [50].
Acetylation is one type of post-translational modification of proteins. The acetylation of the ε-amino group of lysine, which is common, converts a charged side chain to a neutral one. Acetylation/deacetylation of histones also plays a role in gene expression and cancer. These modifications are effected by enzymes called histone acetyltransferases (HATs) and histone deacetylases (HDACs).
Two general mechanisms are known for deacetylation. One mechanism involves zinc binding to the acetyl oxygen. Another family of deacetylases require NAD+, which transfers an ribosyl group to the acetyl oxygen.
https://en.wikipedia.org/wiki/Acetylation
Condensins are large protein complexes that play a central role in chromosome assembly and segregation during mitosis and meiosis (Figure 1). Their subunits were originally identified as major components of mitotic chromosomes assembled in Xenopus egg extracts.
https://en.wikipedia.org/wiki/Condensin
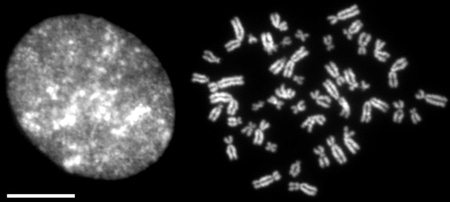
The SARS-CoV-2 envelope (E) protein forms an ion channel, and its C-terminus resembles the N-terminus of histone H3. Therefore, similar to H3, it was shown to directly interact with bromodomains (BRDs) (Table (Table2)2) (Fig. (Fig.2A)2A) [24, 51]. In addition to the interaction of the viral E protein with BRDs, the spike protein of SARS-CoV-2 results in enhancement of BRD4 expression, which is a regulator for senescence mechanism. Therefore, high levels of reactive oxygen species (ROS), DNA damage, and cellular senescence were observed in the infected cell lines. Interestingly, treatment of the cells with a BRD4 inhibitor reversed the senescent phenotype [52].
Several viruses have shown direct interactions with host BRDs as well. For example, the human papillomavirus (HPV) tethers its genome to the host chromosomes through binding of the E2 protein to the host BRD4 [53]. Therefore, tackling the consequences of SARS-CoV-2 binding to bromodomain proteins can lead to promising findings and the discovery of potential viral inhibitors.
Incidentally they don’t refer to MSH3 directly, but they do refer to MutLα and MutSα complexes:
DNA methyltransferase 1 (DNMT1) is an essential player in the process of DNA methylation. Knocking down DNMT1 in telomerase reverse transcriptase (hTERT)-immortalized normal human fibroblasts caused an indirect defect in the MMR pathway, as a consequence of decreased levels of MutLα and MutSα complexes [54].
And a refresher about interactions with tumour suppressors:
The spike (S) glycoprotein of SARS-CoV-2 undergoes proteolytic cleavage at the S1/S2 site by host furin or furin-like proteases [79]. This cleavage results in the surface subunit S1, responsible for the virus attachment to the host cell surface receptor, and the transmembrane subunit S2, which derives the fusion of the viral and host membranes and allows the release of the viral genome into host cells [80]. The interaction of the S2 subunit with p53, BRCA1, and BRCA2 was predicted in silico (Table (Table2)2) (Fig. (Fig.3D)3D) [48]. BRCA1, BRCA2, and p53 are well-known tumor suppressor proteins. BRCA proteins participate in HR to repair DNA DSBs. BRCA1 functions upstream of BRCA2 in response to DNA damage, while BRCA2 plays a main role in the regulation of RAD51 activity in HR machinery [81]. BRCA1 was previously associated with Tat-dependent transcription enhancement of the HIV-1 infection [82]. Moreover, numerous functional activities of BRCA1 are antagonized by interacting with oncogenic HPV E6 and E7 proteins [83]. Further studies are required to confirm the interaction of the proteins in vitro and also to understand the extent of DNA damage caused by this interaction if confirmed.

Experimental evidence for spike protein induced enhanced BRD4 expression
In 2021, Meyer et al conducted an in vitro study using spike protein transfected A549 cells: “SARS-CoV-2 Spike Protein Induces Paracrine Senescence and Leukocyte Adhesion in Endothelial Cells”2.
The A549 cell line consists of hypotriploid alveolar basal epithelial cells. This cell line was first developed by D. J. Giard et al. in 1972 by removing and culturing pulmonary carcinoma tissue from the explanted tumor of a 58-year-old caucasian male.
https://www.synthego.com/a549-cells
Senescence-associated secretory phenotype (SASP) is a phenotype associated with senescent cells wherein those cells secrete high levels of inflammatory cytokines, immune modulators, growth factors, and proteases. SASP may also consist of exosomes and ectosomes containing enzymes, microRNA, DNA fragments, chemokines, and other bioactive factors. Soluble urokinase plasminogen activator surface receptor is part of SASP, and has been used to identify senescent cells for senolytic therapy. Initially, SASP is immunosuppressive (characterized by TGF-β1 and TGF-β3) and profibrotic, but progresses to become proinflammatory (characterized by IL-1β, IL-6 and IL-8) and fibrolytic. SASP is the primary cause of the detrimental effects of senescent cells.
https://en.wikipedia.org/wiki/Senescence-associated_secretory_phenotype
The concept of paracrine senescence (senescence-induced senescence) is essential to understand the impact of the propagation of a senescence response in the tissue microenvironment. Continuous exposure to the SASP induces senescence in bystander normal cells, both in vitro and in vivo (Acosta et al., 2013; Nelson et al., 2012). Therefore, paracrine senescence has been shown to mediate the deleterious effects of SCs on tissue homeostasis, namely in promoting tumourigenesis and aging-related organ dysfunction, as well as in impairing regeneration (Campisi, 2005; Ferreira-Gonzalez et al., 2018; Gonzalez-Meljem et al., 2018).
From the abstract:
NB Synthetic mRNA gene agents also upregulate Il-6, TNFa, Il-1 and other pro-inflammatory cytokines3.
Increased mortality in COVID-19 cases is often associated with microvascular complications. We have recently shown that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein promotes an inflammatory cytokine interleukin 6 (IL-6)/IL-6R-induced trans signaling response and alarmin secretion. Virus-infected or spike-transfected human epithelial cells exhibited an increase in senescence, with a release of senescence-associated secretory phenotype (SASP)-related inflammatory molecules.
A BRD4 inhibitor reverses the senescent effects of spike on epithelial cells:
Introduction of the bromodomain-containing protein 4 (BRD4) inhibitor AZD5153 to senescent epithelial cells reversed this effect and reduced SASP-related inflammatory molecule release in TMNK-1 or EAhy926 (representative human endothelial cell lines), when cells were exposed to cell culture medium (CM) derived from A549 cells expressing SARS-CoV-2 spike protein. Cells also exhibited a senescence phenotype with enhanced p16, p21, and senescence-associated β-galactosidase (SA-β-Gal) expression and triggered SASP pathways.
Evolutionarily, p16 and p21 serve to try to prevent the proliferation of older cells which may potentially have acquired carcinogenic DNA mutations. They are also cancer biomarker proteins:
p21Cip1 (alternatively p21Waf1), also known as cyclin-dependent kinase inhibitor 1 or CDK-interacting protein 1, is a cyclin-dependent kinase inhibitor (CKI) that is capable of inhibiting all cyclin/CDK complexes, though is primarily associated with inhibition of CDK2. p21 represents a major target of p53 activity and thus is associated with linking DNA damage to cell cycle arrest. This protein is encoded by the CDKN1A gene located on chromosome 6 (6p21.2) in humans.
…Cytoplasmic p21 expression can be significantly correlated with lymph node metastasis, distant metastases, advanced TNM stage (a classification of cancer staging that stands for: tumor size, describing nearby lymph nodes, and distant metastasis), depth of invasion and OS (overall survival rate). A study on immunohistochemical markers in malignant thymic epithelial tumors shows that p21 expression has a negatively influenced survival and significantly correlated with WHO (World Health Organization) type B2/B3. When combined with low p27 and high p53, DFS (Disease-Free Survival) decreases.
https://en.wikipedia.org/wiki/P21
p16 (also known as p16INK4a, cyclin-dependent kinase inhibitor 2A, CDKN2A, multiple tumor suppressor 1 and numerous other synonyms), is a protein that slows cell division by slowing the progression of the cell cycle from the G1 phase to the S phase, thereby acting as a tumor suppressor. It is encoded by the CDKN2A gene. A deletion (the omission of a part of the DNA sequence during replication) in this gene can result in insufficient or non-functional p16, accelerating the cell cycle and resulting in many types of cancer.[5][6][7]
p16 can be used as a biomarker to improve the histological diagnostic accuracy of grade 3 cervical intraepithelial neoplasia (CIN). p16 is also implicated in the prevention of melanoma, oropharyngeal squamous cell carcinoma, cervical cancer, vulvar cancer and esophageal cancer.
If you want to accelerate your biological age and accumulate ROS induced DNA damage then spike protein will serve you well:
Inhibition of IL-6 trans signaling by tocilizumab and inhibition of inflammatory receptor signaling by the Bruton’s tyrosine kinase (BTK) inhibitor zanubrutinib, prior to exposure of CM to endothelial cells, inhibited p21 and p16 induction. We also observed an increase in reactive oxygen species (ROS) in A549 spike-transfected and endothelial cells exposed to spike-transfected CM. ROS generation in endothelial cell lines was reduced after treatment with tocilizumab and zanubrutinib.
This is not good at all for your cardiovascular health, and by implication for neurological health:
Cellular senescence was associated with an increased level of the endothelial adhesion molecules vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1), which have in vitro leukocyte attachment potential. Inhibition of senescence or SASP function prevented VCAM-1/ICAM-1 expression and leukocyte attachment. Taken together, we identified that human endothelial cells exposed to cell culture supernatant derived from SARS-CoV-2 spike protein expression displayed cellular senescence markers, leading to enhanced leukocyte adhesion.
The present study addresses effects associated with epithelial cells expressing SARS-CoV-2 spike protein and the paracrine-associated response generated in endothelial cells that leads to senescence. Results from this study identified a mechanism by which SARS-CoV-2 infection propagates the paracrine effect, suggesting potential therapeutic means to interdict relevant mechanistic steps impeding deleterious SASP function.
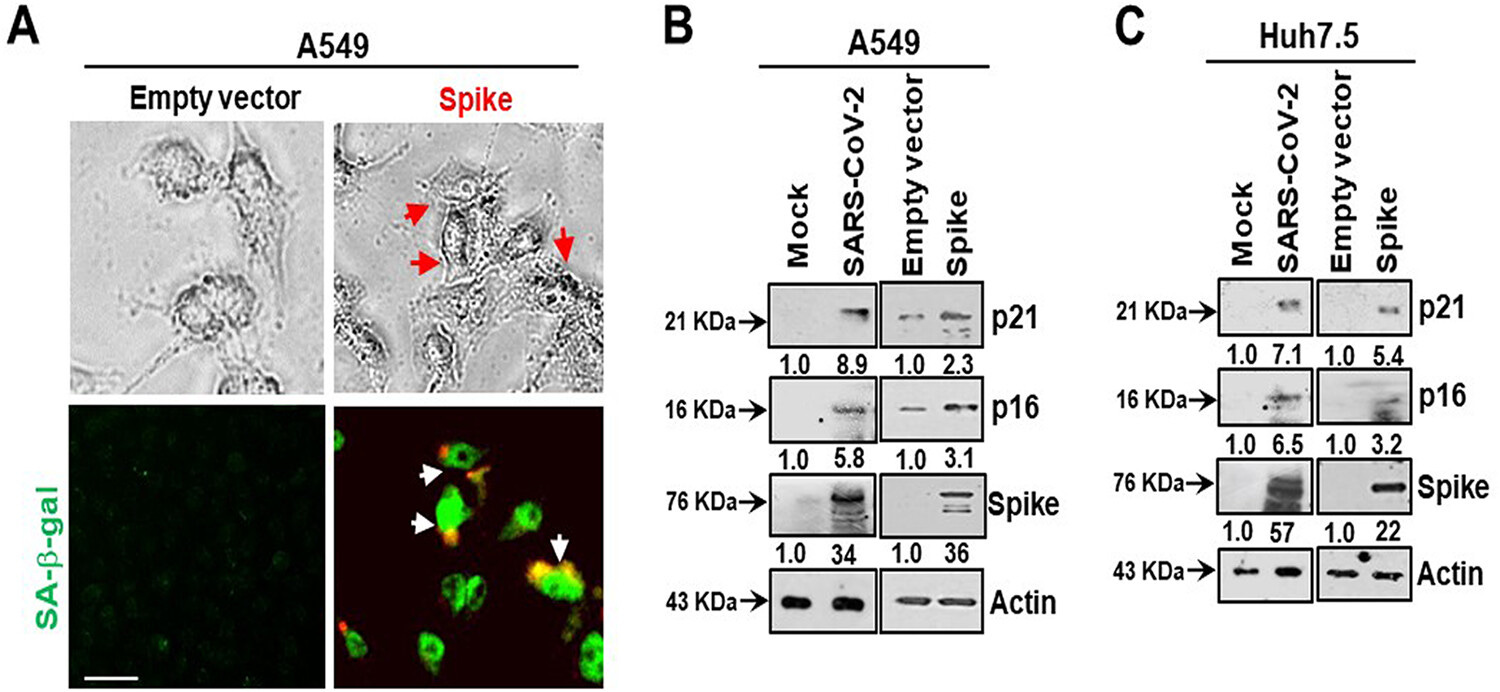
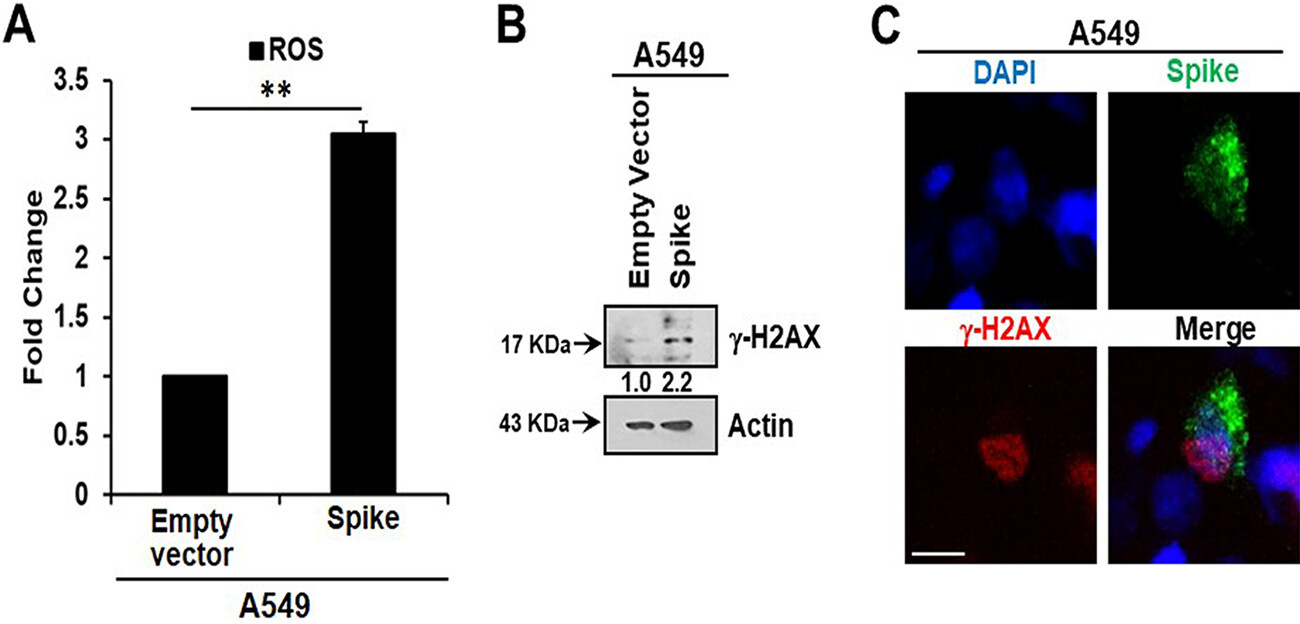
Here, we measured intracellular ROS and observed an approximately 3-fold increase in ROS in A549 spike-transfected cells compared to levels in empty plasmid-transfected control cells (Fig. 2A). Increased production of intracellular ROS contributes to DNA damage, leading to cellular senescence. We observed an increase in the DNA damage response marker γ-H2AX in cells transfected with the viral spike gene (Fig. 2B). γ-H2AX was found to be localized to the nucleus in viral spike-transfected A549 cells (Fig. 2C). Our results indicated that SARS-CoV-2 spike protein expression induces a senescent state in spike-transfected A549 cells that is associated with the DNA damage response and increased ROS generation.
But what about BRD4 and cancer?
Over to wiki again:
Most cases of NUT midline carcinoma involve translocation of the BRD4 gene with NUT genes. BRD4 is often required for expression of Myc and other "tumor driving" oncogenes in hematologic cancers including multiple myeloma, acute myelogenous leukemia and acute lymphoblastic leukaemia.
BRD4 is a major target of BET inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials.
So why are we inducing it via injection en masse whilst other researchers are looking to inhibit the very same protein?
For instance, BRD4 inhibitor AZD5153 represses spike induced BRD4-mediated SASP:
Bromodomain-containing protein 4 (BRD4) is a novel regulator of the senescence mechanism. BRD4 expression is required in senescence immune surveillance and SASP-associated signaling (15). Spike gene transfection of A549 cells led to an enhanced expression of BRD4 (Fig. 3A). A549 cells were transfected with the SARS-CoV-2 spike protein and incubated for 48 h prior to exposure for 16 h to the BRD4 inhibitor AZD5153. Spike protein expression induced p21 and p16 senescence markers, the levels of which were reduced by the addition of AZD5153 (Fig. 3B). Spike-transfected A549 cells exhibited enhanced secretion of SASP-related inflammatory molecules like the alarmin HMGB1 and cytokines interleukin 1α (IL-1α) and IL-6.
It shouldn’t surprise you that AZD5153 is therefore also a tumour suppressor and can suppress the proliferation of CRC cells4.
Paracrine effects mean you don’t need spike protein in the cell to induce the senescence-associated secretory phenotype. This should send shivers down your spine, just like any good horror movie.

Senescent cells secrete inflammatory molecules that may induce paracrine senescence in their surrounding cells. We examined whether senescent epithelial cells generated by spike transfection stimulate paracrine senescence in nearby endothelial cells where SARS-CoV-2 spike protein expression is absent. For this, we incubated endothelial TMNK-1 cells with conditioned culture medium (CM) collected from SARS-CoV-2 spike-expressing A549 cells. We observed green fluorescence associated with SA-β-Gal expression in spike CM-treated cells (Fig. 4A). We also observed a strong expression of the senescence markers p21 and p16 from spike CM-treated TMNK-1 cells.
Release of SASP molecules is a signature of the senescence mechanism. An increase in the levels of SASP release-related inflammatory molecules HMGB1, IL-1α, IL-6, and monocyte chemoattractant protein 1 (MCP-1) was also observed in spike CM-treated TMNK-1 cells (Fig. 4D). Thus, our results indicated an induction of paracrine endothelial cell senescence as a bystander effect from SARS-CoV-2 spike-expressing epithelial cells.

Note that the culture medium prepared with BRD4 inhibitor AZD5153 (no spike) doesn’t express the senescence markers p21 & p16. And look closely and you see an 8 fold increase in IL-6 vs the empty vector control:
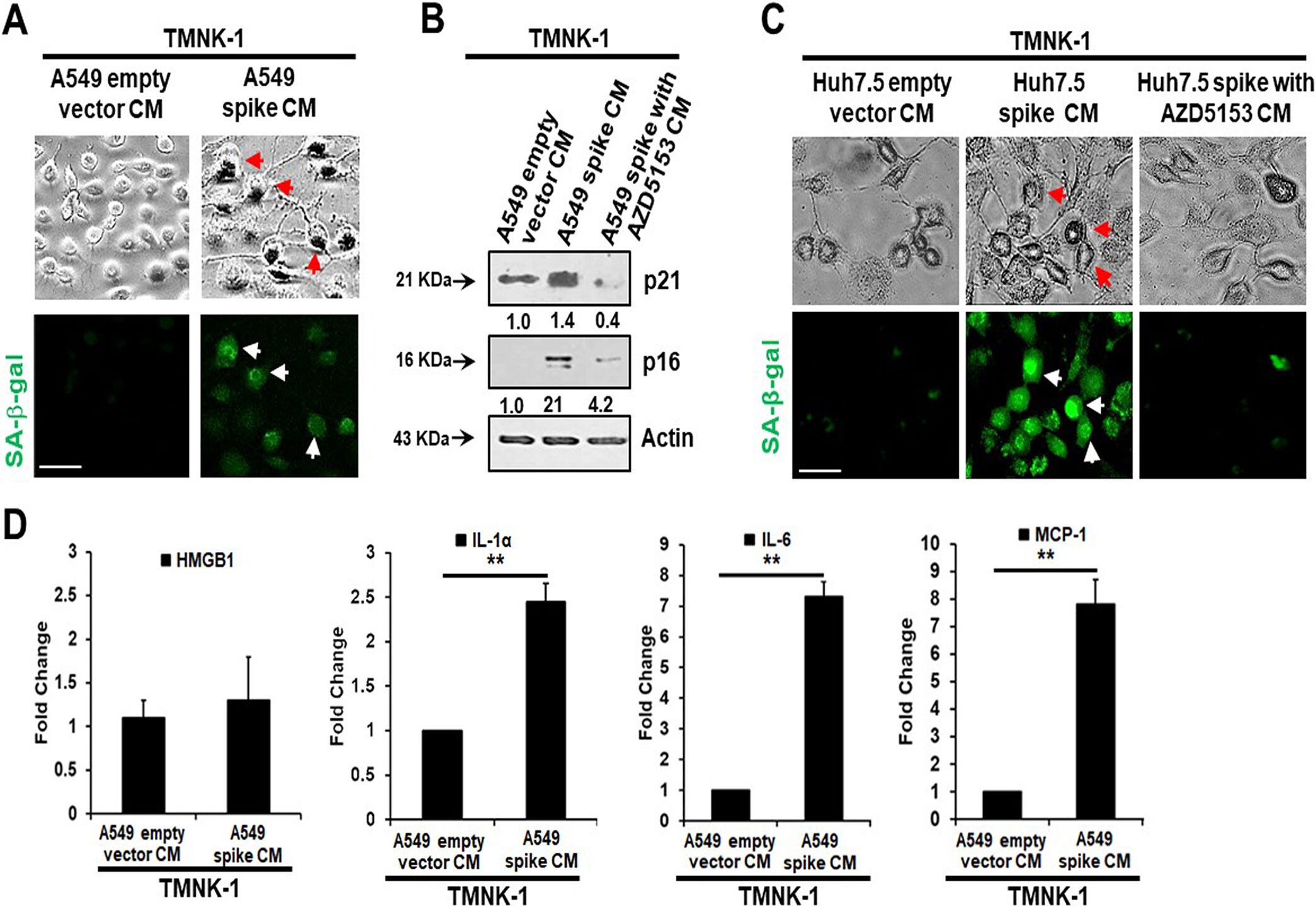
The researchers then tested the hypothesis that pro-inflammatory cytokines and molecules act as paracrine signallers.
It is anticipated that the inflammatory molecules present in CM from spike-expressing cells, such as HMGB1, IL-1α, and IL-6, may bind to different receptors on the endothelial cell surface and stimulate Toll-like receptor (TLR)-mediated signaling. Stimulation of TLR signaling leads to activation of the downstream molecules Akt and p38-MAPK, and these active molecules are crucial regulators of the senescence phenomenon. We observed that exposure of TMNK-1 and EAhy926 endothelial-like cell lines to spike CM resulted in an enhancement in phosphorylation of Akt and p38-MAPK molecules (Fig. 5A and B).
Again, in cancer patients you would absolutely not wish to unnecessarily elevate Il-1, Il6 & TNFa without good reason, or you may be giving a drug on the one hand and reversing its effects on the other.
Would they do this? Of course they would, regardless of whether it worked or not to prevent or transmit infection vs safer, effective alternatives such as ivermectin, which also has powerful anti-cancer properties too5.
Science, see!
And money:

The American Cancer Society (ACS) supports the National Comprehensive Cancer Network (NCCN)* recommendation that all people with cancer should be fully vaccinated against COVID-19.
…The CDC stresses that getting any COVID-19 vaccine is better than being unvaccinated.
The NCCN suggests that people with cancer get one of the mRNA vaccines (Pfizer-BioNTech or Moderna), including for additional doses (see below), if possible. These vaccines are preferred for people with weakened immune systems.
For editorial balance and scientific progress through discussion I include comments from one of the experts in the field (translated):



He very well knows the typical latency periods for solid cancer after initiation. And its rarely 2 years. Plus he doesn’t see the many cases of aggressive recurrence, aka “turbocancer”, which are being presented, aka “evolutionary profiles”:

He also asked “WHERE ARE THE PUBLICATIONS?”
He could start by referring back to an Oncology 101 text book, for example this one, please go to page 190 Jerome:


He could discuss this and argue that despite being associated with tumorigenesis the agent stays in the arm*, isn’t there for long enough etc, but he doesn’t. Its now a binary “yes” or “no” argument without consideration of underlying cancer pathways, just a lot of arm waving and SHOUTING.
This doesn’t help advance any field, and I wouldn’t ask or expect him to prescribe you ivermectin or fenbendazole either, for the same reasons.

* Biodistribution.
LNPs are distributed to most tissues within minutes, at various concentrations678, adenovirus vector vaccines less so9. A more recent citation will be added, if available .

Back to our paper on spike induced paracrine senescence. As an example of a cancer therapeutic which targets inflammatory signalling zanubrutinib is used to treat B-cell lymphomas:
The Bruton’s tyrosine kinase (BTK) inhibitor zanubrutinib is used to treat B-cell malignancies and prevents a vast range of inflammatory receptor signaling, including that by the tumor necrosis factor receptor (TNFR), IRAK, and all TLRs except TLR3 (16). Treatment of TMNK-1 and EAhy926 endothelial cells with zanubrutinib prior to exposure to spike CM caused significant attenuation in Akt and p38-MAPK phosphorylation (Fig. 5A and B).
Unfortunately senescence is a one-way ticket, unless you are happy to lose the cell or risk carcinogenesis.
Neither is a good option, especially if it involves your brain, cardiovascular system, joints or immune system:
Senescence is an irreversible arrest of cell proliferation while the cell maintains metabolic function.

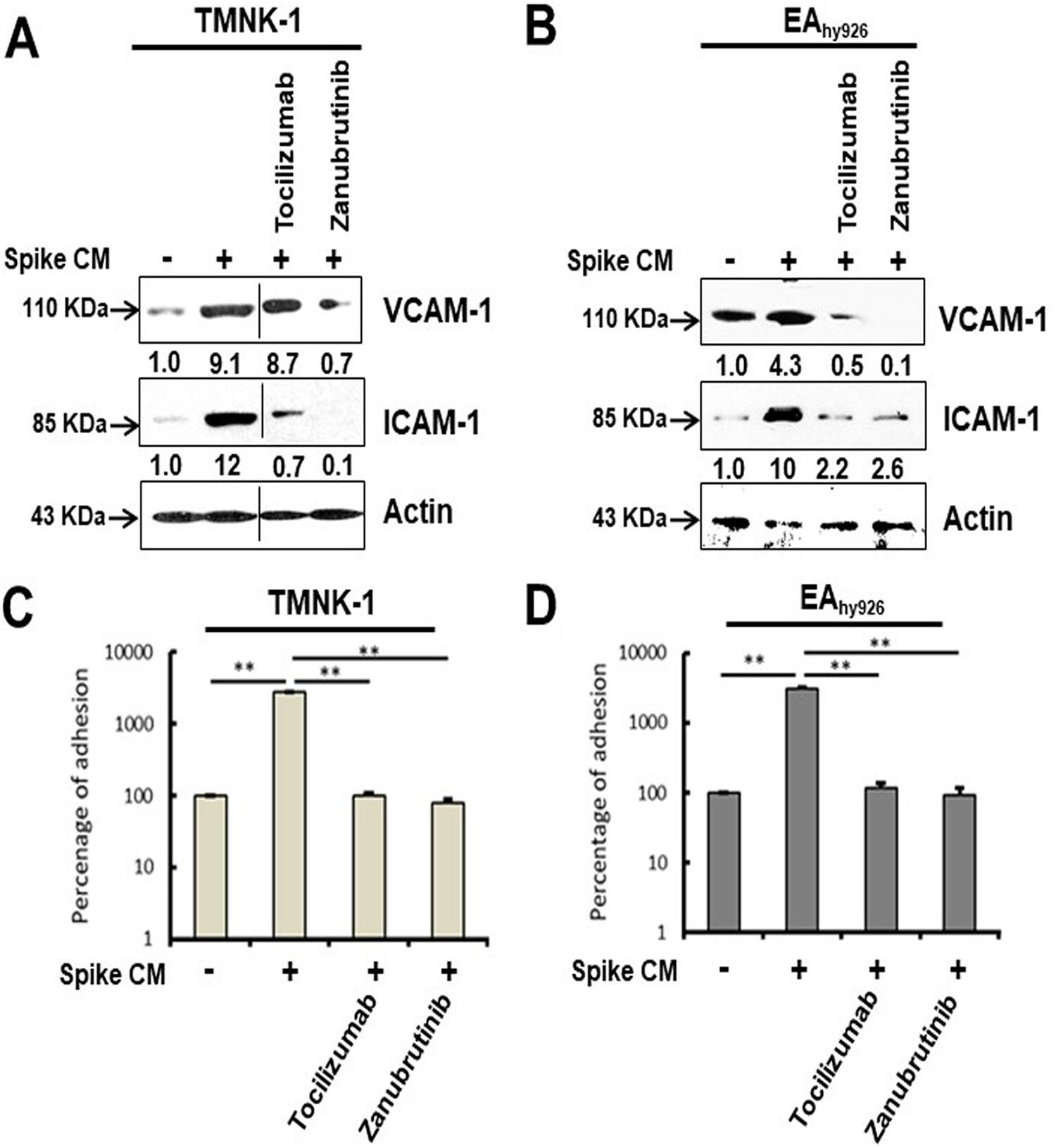
As if BRD4 induced SASP or carcinogenesis isn’t bad enough, endothelial cell surfaces express the adhesion molecules vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) in response to cytokines, contributing to leukocyte attachment and initiating vasculopathy:
Our Western blot analysis showed that both TMNK-1 and EAhy926 cells express increased levels of VCAM-1 and ICAM-1 molecules after exposure to CM from SARS-CoV-2 spike-expressing A549 cells (Fig. 7A and B). These enhanced levels of VCAM-1 and ICAM-1 were reduced after rescue from senescence following treatment with tocilizumab or zanubrutinib. To further determine whether these contrasting phenotypes are seen under physiological conditions, we assessed leukocyte adhesion using a fluorometric assay on the human monocyte cell line THP-1. A significant cellular adhesion of THP-1 cells on the surface of TMNK-1 and EAhy926 cells was detected in the presence of CM from SARS-CoV-2 spike-expressing A549 cells, in association with elevated expression of the adhesion molecules (Fig. 7C and D). The loss of leukocyte adhesion is also corroborated by the reduction of VCAM-1 and ICAM-1 after treatment with tocilizumab or zanubrutinib. These results indicate that leukocyte trafficking occurs to endothelial cells in an in vitro cellular environment, which is influenced by SARS-CoV-2 spike expression and paracrine senescence in surrounding epithelial cells.
Long-term antiretroviral therapy was associated with the decline of VCAM-1 concentration. That may suggest the lowering of endothelial activation and the decreased risk of the development of cardiovascular disease among ARV-treated patients.
…Evidence suggests that VCAM-1 is associated with multiple disorders such as cardiovascular disease, cancer, rheumatoid arthritis, and asthma.
From: “VCAM-1 as a Biomarker of Endothelial Function among HIV-Infected Patients Receiving and Not Receiving Antiretroviral Therapy” (2022).
BRD4, one of the bromodomain and extraterminal (BET) family proteins, plays a vital role in cellular senescence, including stimulation of SASP (18). We observed that inhibition of BRD4 by AZD5153 reduces the release of SASP-related inflammatory molecules and expression of senescence associated marker proteins. A recent study also suggested that BET inhibition blocks inflammation-induced cardiac dysfunction in COVID-19 patients (19).
Quercetin as a senolytic drug, ie it can selectively induce the death of senescent cells to improve health:
ROS are generated as a by-product of cellular metabolism. Accumulation of ROS causes cytostatic effects due to their ability to damage DNA, protein, and lipid molecules in cells. ROS cause a variety of lesions in DNA that lead to DNA double-strand breaks (20). Systemic oxidative stress status is raised in critically ill COVID-19 patients (21). Oxidative stress-mediated DNA damage accelerates cellular senescence (22). Our data suggest that the presence of SARS-CoV-2 spike protein generates ROS in epithelial cells. A clinical trial (ClinicalTrials registration no. NCT04377789) is continuing to assess the effect of the senolytic drug quercetin for prevention and treatment of COVID-19, indicating the perceived importance of senescence in COVID-19 pathogenesis.
Endothelial senescence may lead to microvascular complications by secretion of the cellular adhesion molecules VCAM-1/ICAM-1, which may cause leukocyte adhesion on the surface of the endothelium and may increase coronary blockade.
As per the last Substack, we now know that spike protein expression can persist for over a year after infection, and for several months (if not longer?) after boosting, - and that’s without considering repeat breakthrough infections.
If you are elderly you start out from a high base too:
A nonreplicable form of SARS-CoV-2 spike protein is used in the vaccines for immunization. Therefore, viral spike protein expression and duration of antigenic stimulation to the immune system in the injected tissue (although expected for a brief period) may not be sufficient to exhibit significant senescence or deleterious effect to adjacent endothelial cells. In this scenario, senior or elderly people who already have an accumulation of senescent cells may have a higher possibility of paracrine senescence after vaccination (32).
Related to the above, hat tip to Arkmedic for highlighting a study from 2023 by Zhang et al, investigating the effects of spike protein induced RPE cell senescence and age-related macular degeneration (AMD, ie blindness) using a zebrafish model, which are genetically quite close to humans10.
There are questions to be asked about the study though, such as why it doesn’t refer to the now well established BRD4 induced senescence, let alone the BET family proteins? And they don’t mention the word “\/ a c c i n e” once, which is also curious:
Abstract
SARS-Cov-2 infection, which has caused the COVID-19 global pandemic, triggers cellular senescence. In this study, we investigate the role of the SARS-COV-2 spike protein (S-protein) in regulating the senescence of RPE cells. The results showed that administration or overexpression of S-protein in ARPE-19 decreased cell proliferation with cell cycle arrest at the G1 phase. S-protein increased SA-β-Gal positive ARPE-19 cells with high expression of P53 and P21, senescence-associated inflammatory factors (e.g., IL-1β, IL-6, IL-8, ICAM, and VEGF), and ROS. Elimination of ROS by N-acetyl cysteine (NAC) or knocking down p21 by siRNA diminished S-protein-induced ARPE cell senescence. Both administrated and overexpressed S-protein colocalize with the ER and upregulate ER-stress-associated BIP, CHOP, ATF3, and ATF6 expression. S-protein induced P65 protein nuclear translocation. Inhibition of NF-κB by bay-11-7082 reduced S-protein-mediated expression of senescence-associated factors. Moreover, the intravitreal injection of S-protein upregulates senescence-associated inflammatory factors in the zebrafish retina. In conclusions, the S-protein of SARS-Cov-2 induces cellular senescence of ARPE-19 cells in vitro and the expression of senescence-associated cytokines in zebrafish retina in vivo likely by activating ER stress, ROS, and NF-κb. These results may uncover a potential association between SARS-cov-2 infection and development of AMD.
GP120 induced BRD4 expression
For further experimental evidence I can thank Charles for pointing me in the right direction:
It helped me to uncover an obscure study into HIV-induced neuropathy.
From Takahashi et al (2018), “Spinal bromodomain-containing protein 4 contributes to neuropathic pain induced by HIV glycoprotein 120 with morphine in rats”11.
Unfortunately the full version is paywalled, but key findings are that the gp120 inserts are probably the parts of the spike protein inducing the overexpression, that this may lead to neuropathy, and that taking morphine without BET inhibitors can make the symptoms worse, not better.
What is allodynia? Allodynia is a type of neuropathic pain (nerve pain). People with allodynia are extremely sensitive to touch. Things that don't usually cause pain can be very painful. These may include cold temperatures, brushing hair or wearing a cotton t-shirt.
https://my.clevelandclinic.org/health/symptoms/21570-allodynia
Abstract
The symptoms of HIV-sensory neuropathy are dominated by neuropathic pain. Recent data show that repeated use of opiates enhances the chronic pain states in HIV patients. Limited attention has so far been devoted to exploring the exact pathogenesis of HIV painful disorder and opiate abuse in vivo, for which there is no effective treatment. Bromodomain-containing protein 4 (Brd4) is a member of the bromodomain and extraterminal domain protein (BET) family and functions as a chromatin ‘reader’ that binds acetylated lysines in histones in brain neurons to mediate the transcriptional regulation underlying learning and memory. Here, we established a neuropathic pain model of interaction of intrathecal HIV envelope glycoprotein 120 (gp120) and chronic morphine in rats. The combination of gp120 and morphine (gp120/M, for 5 days) induced persistent mechanical allodynia compared with either gp120 or morphine alone. Mechanical allodynia reached the lowest values at day 10 from gp120/M application, beginning to recover from day 21. In the model, gp120/M induced overexpression of Brd4 mRNA and protein at day 10 using RT-qPCR and western blots, respectively. Immunohistochemical studies showed that Brd4 at day 10 was expressed in the neurons of spinal cord dorsal horn. BET inhibitor I-BET762 dose-dependently increased the mechanical threshold in the gp120/M pain state. The present study provides preclinical evidence for treating HIV neuropathic pain with opioids using the BET inhibitor.
BRD4 and cancer
Zhu et al (2017) used a bromodomain and extraterminal protein inhibitor called JQ1 to suppress thyroid tumour growth in a mouse model12.
Myc is a family of regulator genes and proto-oncogenes that code for transcription factors. The Myc family consists of three related human genes: c-myc (MYC), l-myc (MYCL), and n-myc (MYCN). c-myc (also sometimes referred to as MYC) was the first gene to be discovered in this family, due to homology with the viral gene v-myc.
In cancer, c-myc is often constitutively (persistently) expressed. This leads to the increased expression of many genes, some of which are involved in cell proliferation, contributing to the formation of cancer. A common human translocation involving c-myc is critical to the development of most cases of Burkitt lymphoma. Constitutive upregulation of Myc genes have also been observed in carcinoma of the cervix, colon, breast, lung and stomach.
Myc is thus viewed as a promising target for anti-cancer drugs.
Results: JQ1 markedly inhibited thyroid tumor growth and prolonged survival of these mice. Global differential gene expression analysis showed that JQ1 suppressed the cMyc (hereafter referred to as Myc) transcription program by inhibiting mRNA expression of Myc, ccnd1, and other related genes. JQ1-suppressed Myc expression was accompanied by chromatin remodeling as evidenced by increased expression of histones and hexamethylene bis-acetamide inducible 1, a suppressor of RNA polymerase II transcription elongation. Analyses showed that JQ1 decreased MYC abundance in thyroid tumors and attenuated the cyclin D1–CDK4–Rb–E2F3 signaling to decrease tumor growth. Further analysis indicated that JQ1 inhibited the recruitment of BDR4 [sic] to the promoter complex of the Myc and Ccnd1 genes in rat thyroid follicular PCCL3 cells, resulting in decreased MYC expression at the mRNA and protein levels to inhibit tumor cell proliferation.
And from 2013, Floyd et al investigated the specific mechanisms behind the inhibition of DDR by BRD413:
We further investigated the role of chromatin structure in the DDR by monitoring ionizing radiation-induced signaling and response events with a high-content multiplex RNAi screen of chromatin modifying and interacting genes. We discovered that an isoform of Brd4, a bromodomain and extra-terminal (BET) family member, functions as an endogenous inhibitor of DDR signaling by recruiting the condensin II chromatin remodeling complex to acetylated histones via bromodomain interactions. Loss of this isoform results in relaxed chromatin structure, rapid cell cycle checkpoint recovery and enhanced survival post-irradiation, while functional gain of this isoform compacted chromatin, attenuated DDR signaling, and enhanced radiation-induced lethality. These data implicate Brd4, previously known for its role in transcriptional control, as an insulator of chromatin that can modulate the signaling response to DNA damage.
An article by Zhang et al also discusses BRD2, ACE2 and conflicting viewpoints.
From “Targeting ACE2-BRD4 in Cancer and COVID-19” (2023)14:
Bromodomain and extraterminal domain (BET) inhibitors target the ‘readers’ of acetylated lysine residues on histone and non-histone proteins (9). In the context of cancer therapy, BET inhibitors downregulate MYC and other oncogenic factors (10-12). However, recent reports provided conflicting viewpoints regarding the use of pan-BET inhibitors in targeting ACE2 overexpression. In lung cancer cells, the BET protein BRD2 was identified as 'proviral' and positively regulated expression of the ACE2 receptor, whereas BRD4 was ‘antiviral’ through changes in gene expression profiles, including interferon signaling (13). Thus, anticancer agents targeting both BRD2 and BRD4 might be disadvantageous in the setting of COVID-19 infection.
High expression of ACE2 and BRD4 mRNAs is associated with increased mortality if you have CRC:
Colorectal cancer datasets predicted that high expression of both ACE2 and BRD4 mRNAs was associated with significantly reduced patient survival compared with low ACE2 plus low BRD4 levels (14,15). No such relationships were observed for ACE2 and BRD2. In a panel of human colon cancer cells, there was moderate to high constitutive expression of ACE2, BRD2 and BRD4 proteins compared with normal colonic epithelial cells. RNAi-mediated knockdown of ACE2 or loss of ACE2 upon pharmacologic BET inhibition with JQ1, MZ1 or dBET6 (12,16,17) increased DNA damage/repair markers, reduced cell viability/colony formation, and enhanced apoptosis. Morphological and mechanistic readouts were reversed following transient transfection of an ACE2 expression construct, which dose-dependently circumvented the apoptosis induction associated with decreased BRD4 levels upon dBET6 treatment. Antitumor activity of dBET6 in vivo coincided with a near-complete loss of tumor-associated BRD4 and ACE2 protein expression (12,15).
In other words, if you have elevated BRD4 then ACE2 may assume an oncogenic role due to crosstalk with BRD4 and c-Myc:
Our findings and prior reports highlight the yin-yang nature of targeting ACE2 and BET proteins in cancer etiology. In human colon cancer cells, an apparent oncogenic role was implicated for ACE2 via crosstalk with BRD4 and c-Myc (15). In human lung epithelial cells, BET antagonists inhibited SARS-CoV-2 infection (18), whereas in lung cancer cells proviral versus antiviral roles were identified for BRD2 and BRD4, respectively (13). Next-generation therapeutics that selectively target BRD2 or BRD4 will help to clarify risk-benefit in the context of prior versus concurrent BET inhibitor treatment in colorectal cancer patients with SARS-CoV-2 infection.
Tsukamoto et al (2020) conducted an in vitro investigation into how the inhibition of BRD4 can be used in a targeted therapeutic approach for the treatment of mantle cell lymphoma15.
Mantle Cell lymphoma (MCL) is an aggressive, rare form of non-Hodgkin lymphoma (NHL) that arises from cells originating in the “mantle zone.” The mantle zone is the outer ring of small lymphocytes surrounding the center of a lymphatic nodule. MCL accounts for roughly six percent of all NHL cases in the United States.
https://lymphoma.org/understanding-lymphoma/aboutlymphoma/nhl/mantle-cell-lymphoma/
Background: Since bromodomain-containing protein 4 (BRD4) facilitates the transcription of genes important for neoplastic cells in a cancer-type specific manner, BRD4-regulated molecules may also include therapeutic targets for mantle cell lymphoma (MCL), a treatment-refractory subtype of malignant lymphoma.
Materials and Methods: In order to uncover direct BRD4-regulated targets in MCL, we performed integrated analysis using the pathway database and the results of both gene-expression profiling and chromatin immunoprecipitation with parallel sequencing for BRD4.
Results: Treatment with BRD4 inhibitor I-BET151 exerted a dose-dependent inhibitory effect on cell proliferation in MCL cell lines. BRD4 was found to directly regulate series of genes involved in the B-cell receptor (BCR) signaling pathway, including B-cell linker (BLNK), paired box 5 (PAX5), and IKAROS family zinc finger 3 (IKZF3), and several oncogenes, such as MYB. Indeed, the combinatory inhibition of BCR pathway and IKZF showed an additive antitumor effect. Conclusion: Concomitant targeting multiple BRD4-regulated molecules may constitute a rational therapeutic strategy for MCL.
BRD4 can also protect lymphoma cells from ferroptosis, which forms an important part of our innate anti-tumour immunity and immune microenvironment16.
Schmitt et al (2023) investigated the link between BRD4, ferroptosis and diffuse large B-cell lymphoma17. Unfortunately the full paper is paywalled:
Diffuse large B-cell lymphoma (DLBCL), the most common form of non-Hodgkin lymphoma, is characterized by an aggressive clinical course. In approximately one-third of DLBCL patients, first-line multi-agent immunochemotherapy fails to produce a durable response. Molecular heterogeneity and apoptosis resistance pose major therapeutic challenges in DLBCL treatment. To circumvent apoptosis resistance, the induction of ferroptosis might represent a promising strategy for lymphoma therapy. Here, a compound library targeting epigenetic modulators was screened to identify ferroptosis-sensitizing drugs.
I always like to see words like “strikingly” in a paper:
Strikingly, bromodomain and extra-terminal domain (BET) inhibitors sensitized cells of the germinal center B cell-like (GCB) subtype of DLBCL to ferroptosis induction and the combination of BET inhibitors with ferroptosis inducers, such as dimethyl fumarate (DMF) or RSL3, synergized in the killing of DLBCL cells in vitro and in vivo. On the molecular level, the BET protein BRD4 was found to be an essential regulator of ferroptosis suppressor protein 1 (FSP1) expression and to thus protect GCB-DLBCL cells from ferroptosis.
Its 2023 and they aren’t using BRD4 inhibitors with ferroptosis-inducing agents in treatment protocols:
Collectively, we identified and characterized BRD4 as an important player in ferroptosis suppression in GCB-DLBCL and provide a rationale for the combination of BET inhibitors with ferroptosis-inducing agents as a novel therapeutic approach for DLBCL treatment.
I would start with lactoferrin, but that’s just me:
Rationale: Iron-saturated Lf (Holo-Lactoferrin, Holo-Lf) exhibits a superior anticancer property than low iron-saturated Lf (Apo-Lf). Ferroptosis is an iron-dependent cell death characterized by the accumulation of lipid peroxidation products and lethal reactive oxygen species (ROS).
Methods: The effect of different iron-saturated Lf on ferroptosis and radiotherapy were tested on triple-negative breast cancer (TNBC) cell line MDA-MB-231 and non-TNBC cell line MCF-7.
Results: Holo-Lf significantly increased the total iron content, promoted ROS generation, increased lipid peroxidation end product, malondialdehyde (MDA), and enhanced ferroptosis of MDA-MB-231 cells.
From: “Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer” (2021)
Related to the above research, we have from 2021: “Rapid Progression of Angioimmunoblastic T Cell Lymphoma Following BNT162b2 mRNA Vaccine Booster Shot: A Case Report”18:
The remarkable efficiency of nucleoside-modified SARS-CoV-2 mRNA vaccines has been related to their ability to induce a potent stimulation of T follicular helper (TFH) cells, resulting in persistent germinal center B cell responses (1, 2). Clinically, this might translate into reactive lymphoadenopathy which sometimes may raise a differential diagnosis with a lymphoproliferative disorder (3, 4). At the same time, the possible impact of SARS-CoV-2 mRNA vaccination on pre-existing peripheral T cell lymphoma is still to be determined.
A 66-year-old man with no significant medical history except for hypertension, hypercholesterolemia and type 2 diabetes presented on September 1, 2021 with cervical lymphadenopathies that became recently apparent during a flu-like syndrome. The two doses of BNT162b2 mRNA vaccine had been administered, respectively, 5 and 6 months earlier in the left deltoid. Besides moderate asthenia, he did not report any constitutional symptom. Blood examination indicated a mild inflammatory syndrome, without anemia or white blood cell changes; Lymphocytes immunophenotyping was unremarkable. Protein electrophoresis and immunoglobulin levels were normal and Coombs test was negative.
Stage 4 means the cancer has spread from where it started to another body organ. For example to the liver or lung. This is also called secondary or metastatic cancer.
https://www.cancerresearchuk.org/about-cancer/what-is-cancer/stages-of-cancer
A 18F-FDG PET/CT revealed multiple voluminous hypermetabolic lymphadenopathies above and below the diaphragm as well as several extra-nodal hypermetabolic lesions (Figure 1, left panel). Considering a presumptive diagnosis of stage IV lymphoma, a left cervical lymph node biopsy was performed. Pathological examination revealed residual atrophic germinal centers, surrounded by an expanded paracortical area composed of an atypical T-cell infiltrate with clear cell morphology, expressing TFH cell markers (CD3, CD4, PD1, ICOS, BCL6, CXCL13) and a loss of CD7. The paracortical area contained an increased number of high-endothelial venules, supported by an increased number of follicular dendritic cell networks, with some foci of EBV+ B-cell immunoblastic proliferation in the background (Figure 2). These features highly suggested a diagnosis of AngioImmunoblastic T cell Lymphoma (AITL), pattern 2. Next generation sequencing (NGS) performed on the biopsy specimen identified the RHOA G17V mutation characteristic of AITL (5) together with the DNMT3A, IDH2 and TET2 mutations. A TCR-gamma gene rearrangement confirmed a clonal T cell proliferation. Altogether, these findings unambiguously established the diagnosis of AITL. A bone marrow biopsy did not reveal neither morphological nor phenotypic abnormalities, but NGS revealed DMNT3A and TET2 mutations in bone marrow cells with allele frequencies of 41% and 36%, respectively.

Fourteen days after the PET/CT, a booster dose of the BNT162b2 mRNA vaccine was administered in the right deltoid in preparation of the first cycle of chemotherapy. Within a few days following the vaccine booster, the patient reported noticeable swelling of right cervical lymph nodes. In order to get a baseline close to the initiation of the therapy, a second 18F-FDG PET/CT was performed 8 days after the vaccine booster administration, i.e. 22 days after the first one.
This wasn’t just stimulating the TFH cells, it was tumorigenic in action:
It demonstrated a clear increase in number, size and metabolic activity of pre-existing lymphadenopathies at the supra- and sub-diaphragmatic level. Furthermore, new hypermetabolic lymphadenopathies and new hypermetabolic sites had developed since the first examination, in several different locations (Figure 1, right panel).
Total lesion glycolysis (TLG), which measures metabolic activity, was used to monitor changes in lymph node activities:
As compared with the initial test, there was a marked 5.3-fold increase in whole-body TLG, with the increase in the post-booster test being twice higher in the right axillary region than in the left one. In parallel, a mild increase in blood levels of ferritin, C-reactive protein and LDH were noted.
Jerome and his 50 colleagues might be happy with this, but I wouldn’t be and this isn’t just 10 random cases with a weak correlation.
Although not yet full-on disease its a huge red flag, a precursor signal:
Published studies on hypermetabolic lymphadenopathy after SARS-CoV-2 vaccination were recently reviewed and the subject of a meta-analysis (8, 12). Most observations were reported after injection of approved nucleoside-modified mRNA vaccines, namely BNT162b2 (Pfizer-BioNTech) or mRNA-173 (Moderna) (8). Nevertheless, hypermetabolic lymphadenopathies were also observed in 31 health workers following injection of the adenovirus-vectored Vaxveria vaccine (13).
This is totally unacceptable:
Considering oncologic patients, the most informative study was conducted in a series of 728 patients having received the BNT162b2 mRNA vaccine (14). PET/CT revealed hypermetabolic lymph nodes in the axillary and supraclavicular regions draining the vaccine injection site in 36% of the subjects having received the first dose and 54% of those studied after the 2nd dose. The hypermetabolic lymph nodes were enlarged in 7% of 1st dose vaccinees and 18% of 2nd dose vaccinees. Both differences were statistically significant, demonstrating that the impact on draining lymph nodes was greater after the booster dose, confirming data from the meta-analysis above (12). Regarding the relationship with the underlying malignancy, hypermetabolic lymph nodes were considered as malignant in 5% of the patients while no conclusion regarding the malignant nature could be drawn in 15% of the vaccinees including 16 patients with lymphoma.
Even the authors are perplexed:
Interestingly, in none of these studies, the possibility that the mRNA vaccines could have played a role in the development of malignant lymph nodes was considered. Indeed, the consensus so far is that the occurrence of hypermetabolic lymphadenopathies should not question the safety of mRNA vaccines, neither in healthy individuals nor in patients with neoplastic conditions (15).

To the best of our knowledge, this is the first observation suggesting that administration of a SARS-CoV-2 vaccine might induce AITL progression. Several arguments support this possibility. First, the dramatic speed and magnitude of the progression manifested on two 18F-FDG PET-CT performed 22 days apart. Such a rapid evolution would be highly unexpected in the natural course in the disease. Since mRNA vaccination is known to induce enlargement and hypermetabolic activity of draining lymph nodes, it is reasonable to postulate that it was the trigger of the changes observed. Indeed, the increase in size and metabolic activity was higher in axillary lymph nodes draining the site of vaccine injection as compared to their contralateral counterparts. However, pre-existing lymphomatous nodes were also clearly enhanced as compared to the first test. Moreover, new hypermetabolic lesions most likely of lymphomatous nature clearly appeared at distance of the injection site.

Indeed, TFH cells are key targets of these synthetic mRNA gene transfection agents. If you have pre-existing mutations then you are particularly at risk:
In fact, the supposed enhancing action of the vaccine on AITL neoplastic cells is fully consistent with previous observations identifying TFH cells within germinal centers as key targets of nucleoside-modified mRNA vaccines both in animals and in man (1, 2). Malignant TFH cells, the hallmark of AITH, might be especially sensitive to mRNA vaccines when they harbor the RHOA G17V mutation which was present in our case. Indeed, this mutation facilitates proliferation and activation of several signaling pathways in TFH cells (16). Furthermore, mice genetically engineered to reproduce the RHOA G17V and TET2 mutations—both were present in our case—develop lymphoma upon immunization with sheep red blood cells (16). This experimental observation is relevant to RNA vaccines as RNA of sheep red blood cells was shown to be responsible for their ability to stimulate TFH and induce germinal center reaction (17).
No-one was listening:
Our case first raises the question of the COVID-19 prevention strategy to be used in this patient which is currently poorly protected against COVID-19. On the short term, the only option is to recommend strict masking and social distancing, and to offer him anti-SARS-CoV-2 antibody therapy in case of high-risk contact (16). On the longer term, the use of mRNA vaccines should clearly be avoided while other types of vaccines might be considered.
As to latency periods, or how long before we would expect to see a marked increase in cases over and above what we see now, we are looking at an average of 4-5 years for blood-related cancers and 9 years for solid tumours, but there is wide variation:
The latency period can vary greatly with different carcinogens, but even with a single carcinogen, there can be variations in both the latency period and the type of cancers which arise. A 2017 study looked at secondary cancers in people with acute leukemia. We know that chemotherapy—while it can sometimes cure these cancers—can also be a carcinogen which causes other cancers down the line.
Conducted in Argentina, the study followed people with acute leukemias or lymphomas to determine both the incidence of secondary cancers (cancers caused by cancer treatments) and the average latency period between the treatment of the original leukemia or lymphoma and the development of secondary cancer. Roughly one percent of survivors developed secondary cancer. The latency period was significantly shorter for secondary blood-related cancers than for solid tumors. The average latency period for hematologic (blood-related) cancers such as leukemias and lymphomas was 51 months but varied from 10 to 110 months. The average latency period for solid tumors was 110 months, but with this period of time ranging from 25 to 236 months.
https://www.verywellhealth.com/what-is-cancer-latency-period-4057124
BRD4, neurological disorders and cardiovascular pathologies
BET proteins can contribute to neuroinflammation, as per Martella et al (2023)19.
We also saw patents for MSH3 suppressive treatments in the last Substack relating to this.
BET proteins: Bromodomain and extra-terminal proteins.
In recent years, thanks to the development of BET inhibitors, interest in this protein family has risen for its relevance in brain development and function. For example, experimental evidence has shown that BET modulation affects neuronal activity and the expression of genes involved in learning and memory. In addition, BET inhibition strongly suppresses molecular pathways related to neuroinflammation. These observations suggest that BET modulation may play a critical role in the onset and during the development of diverse neurodegenerative and neuropsychiatric disorders, such as Alzheimer’s disease, fragile X syndrome, and Rett syndrome.

Though incomplete, some evidence has shed new light into the molecular pathways linking BRD4 and neuronal activity (Figure 2). Indeed, neurotransmitter and neurotrophic factors induce signaling cascades leading to the activation of PKA and Casein Kinase 2 (CK2). These kinases, in turn, phosphorylate BRD4, favoring its binding to acetylated histones [15,45,66].


Again, disruption of BET proteins up or down may induce all manner of pathologies, the authors note that this work deserves further investigation:
Another study documented elevated abundance of Brd4 mRNA in rat striatal neurons, where BRD4 is involved in dopamine-induced and cAMP/PKA-dependent basal transcription. Indeed, cAMP/PKA signaling elicits BRD4 recruitment to genes induced by dopamine stimulation, whereas pharmacological or genetic inactivation of BRD4 significantly downregulates the transcription of a subset of genes mediated by D1R [66]. Consistent with the role of BRD4 in dopaminergic signaling, Brd4 transcript levels are much higher than Brd2 and Brd3 in nucleus accumbens (NAc), a region enriched in D1R- and D2R-expressing neurons implicated in reward mechanisms [67]. This finding is further supported by the reportedly high concentration of BET proteins in the amygdala and midbrain, also involved in reward behavior in both rodents and non-human primates [52]. Other remarkable differences in the expression of specific BETs have been detected in some neuronal subtypes. For instance, Brd2 expression is prevalent in cerebellar neurons, while the highest Brd4 levels were found in claustral neurons of the frontal cortex and in some hippocampal neurons [65].
Li and colleagues demonstrated that inhibition of BET proteins by JQ1 promotes differentiation of mouse cortical neural progenitor cells (NPCs) towards a neuronal, rather than a glial, phenotype. In particular, genes related to cell cycle progression and glial differentiation were decreased, whereas pro-neurogenic gene expression was sustained. Consistently, BRD2, BRD3, and BRD4 levels decreased, indicating that their suppression is directly associated with neuronal differentiation [75].
Among BET family members, BRD2 seems a crucial regulator strongly implicated in neurodevelopment, in relation to its wide expression in the developing forebrain, midbrain, hindbrain, and spinal cord [77]. Brd2-deficient mice showed early embryonic lethality, as well as generalized developmental and growth delays with impaired neurulation and consequent hindbrain exencephaly [53]. Embryos carrying a deletion in the Brd2 gene showed defects in neural tube closure at the cranial level and craniofacial malformations with irregular thickenings in the neuroepithelial layers of the rostral hindbrain [77]. These data indicate that BRD2 is required for proper neural tube closure, suggesting an essential role of this factor in regional specification of the developing hindbrain.
Administering BRD4 promotors during pregnancy without long term trial safety data risks leading to teratogenic consequences, once again:
Given the involvement of BET proteins in a plethora of processes that promote, maintain, and modulate neurodevelopment, it is not surprising that BRD4 has also been assigned to key functions in embryonic development. Recently, a novel role for BRD4 in the differentiation of mouse neural crest cells has been revealed. Indeed, embryos with neural crest-specific deletion of Brd4 showed skeletal dysplasia, craniofacial defects, and cleft palate, as well as complex cardiac anomalies and consequent perinatal lethality [81]. Furthermore, Brd4 knockout in mice leads to embryonic death in the early post-implantation stages [82].
Consistently, Brd4 knockout in the developing cerebellum resulted in aberrant cytoarchitecture, with the formation of the cerebellar layers that persisted into postnatal development; the reduced size of the cerebellum correlated with behavioral deficits in mice, which displayed symptoms of cerebellar ataxia. Taken together, these data demonstrate that BRD4 is an essential regulator of GCP proliferation and cerebellar development in vivo [84].
BET inhibition impairs the extinction of auditory fear memory but does not alter the acquisition of contextual fear conditioning, lithium chloride-conditioned place aversion, or Barner maze or Y-maze test. Fear extinction is commonly used to treat anxiety disorders such as post-traumatic stress disorder (PTSD) and specific phobias. During fear extinction, excess fear is suppressed by re-exposure to the fear-triggering stimulus in the absence of any aversive event [94,95,96].
Brd4 KO mice exhibited an increase in fear response during both the extinction training and extinction test compared to control mice. However, JQ1-treated mice presented more fear-related behaviors (e.g., freezing) during the extinction test phase compared to the control group. The seeming discrepancy of these results may be explained by total abolishment of BRD4 activity in the knockout mouse vs. its only partial suppression following JQ1 treatment [100].
The effects of BET inhibition on behavior are complex and occasionally contradictory among studies. Despite some studies suggesting that BET inhibition disrupts memory and induces autism-like behavioral deficits, others reported memory improvement in wild-type animals and the occurrence of beneficial effects in mouse models of neurological disorders [66]. To deeply understand the nuances of BET function, it will be necessary to elucidate the mechanisms of BET regulation in different brain regions and in response to various neuronal signaling pathways.

I listed COX-2 inhibitors in the last Substack:
In the nervous tissue, microglia are key players in the regulation of innate immune response and neuroinflammation [125,126]. Specifically, recent data demonstrate that enhanced degradation of BET proteins achieved by PROteolysis-TArgeting Chimera (PROTAC) technology blunted the LPS-induced pro-inflammatory response in the murine microglial cell line SIM-A9. In detail, treatment with dBET1 induced a significant degradation of BRD2 and BRD4, which was associated with a considerable reduction in iNOS and COX-2 levels. Furthermore, BET degradation attenuated the expression of different pro-inflammatory genes, such as Nos2, Ptgs2, Il-1β, Tnfα, Ccl2, Il-6, and Mmp9.
Implications for recovery from ischaemic stroke:
Recent studies employed models of permanent and transient cerebral ischemia to evaluate the effects of BET inhibition on the inflammatory response mediated by NF-kB [131,132,133,134]. In rats, transient cerebral ischemia obtained by middle cerebral artery occlusion (MCAO) resulted in a significant increase in BRD4 expression in the MCAO group compared to the control group, which was prevented by JQ1 treatment. JQ1 administration also hindered the infarct volume, reduced the number of apoptotic cells, and decreased the expression of pro-inflammatory factors (IL-1β, IL-6, IL-17, and TNF-α) in the ischemic brain. Notably, BET blockade suppressed neuroinflammation by reducing p65 levels and by increasing the cytosolic expression of the NF-kB inhibitor IkB [131]. Other reports corroborate these findings, demonstrating that NF-kB suppression by BET blockade markedly hampered the expression of pro-inflammatory mediators, leading to the reduction in pyroptosis and inflammasome activation. From a functional point of view, the molecular changes promoted by BET inhibition were associated with a partial recovery of neurological deficits [132].
BET inhibitors can be neuroprotective:
Neuroprotection mediated by BET inhibition was also confirmed in mouse models of permanent cerebral ischemia. For instance, BRD4 blockade by using dBET1, a proteolysis-targeting chimera, attenuated the infarct volume in permanent focal cerebral ischemia, and this was related to a decrease in pro-inflammatory mediators such as TNF-α, CXCL1, CXCL10, CCL2, and matrix metalloproteinase-9 (MMP-9). dBET1 administration significantly prevented BBB abnormalities, as well as neutrophil infiltration into the ischemic area. Interestingly, dBET1 significantly ameliorated the neurological symptomatology [134]. Reduction in inflammation and preserved BBB integrity following BET inhibition was also reported in the C57BL/6J mouse model subjected to transient MCAO. Indeed, dBET1 reduced pro-inflammatory cytokines, neutrophil infiltration, MMP-9 protein levels, and infarct volume. The protective effects of dBET1 against ischemic damage are attributable not only to its anti-inflammatory function, but also to its anti-oxidant function.
Stroke associated fibrotic scarring may also be inhibited by JQ1:
An additional function attributable to BET proteins in stroke-associated neuroinflammation has emerged from a recent study focusing the attention on the formation of fibrotic scars. It has been shown that TGF-β1 stimulation increased BRD4 expression in a cell culture model of fibrosis and simultaneously promoted fibroblast proliferation and migration. In contrast, JQ1 blocked these effects. The results obtained from in vitro studies were further supported by in vivo models. Specifically, rats underwent transient MCAO developed fibrosis in the infarcted area, which was attenuated when BRD4 was knocked down by adenovirus. Concurrently, ischemic damage, infarct volume, and cognitive alterations appeared to be reduced [137].
BET inhibitors may also act therapeutically to treat neurodegenerative diseases such as AD, Parkinson’s disease (PD), Huntington disease (HD), amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig's Disease, a type of motor neuron disease), or frontotemporal dementia (FTD):
…emerging evidence suggests that BET inhibitors could be valuable targets to treat AD and other neurodegenerative diseases [65,142]. Experimental findings demonstrated the beneficial effects of BET blockade in both wild type and AD mouse models; JQ1 treatment showed an improvement in spatial and associative memory, as well as in long-term potentiation (LTP).
In conclusion:
Since BET proteins are emerging as crucial epigenetic players in CNS physiology, it is not surprising that alterations in their activity may be associated with several neuropathological conditions. From this perspective, the use of BET inhibitors in preclinical research facilitated the dissection of physiopathological mechanisms in cell culture and animal disease models, providing the rationale to target BET proteins as a novel therapeutic strategy.
Collectively, these findings help to shed light on BET proteins’ activity and their possible inhibition when approaching disease treatment. However, several issues need to be addressed. As already reported, more efforts should be made in terms of fundamental research to deeply dissect the role of each BET protein in brain cells; a better comprehension of the molecular, cellular, and functional processes is indeed essential to properly comprehend the contribution of BET proteins in CNS physiopathology and the possible employment of BET inhibition as effective therapeutic strategy. In this context, it will also be important to assess the pharmacological efficacy of the selective manipulation of a specific BrD. Indeed, BrD1- or BrD2-selective BET inhibitors have been recently developed and showed encouraging therapeutic effects with less adverse events [201].
Unfortunately, BRD4 stimulation of fibroblasts can also occur in the myocardium too. I have been warning of the risks of myocardial fibrosis since 2021, along with accelerated atherosclerosis through multiple pathways. And clinical data shows that it may take 10 years before it becomes symptomatic after radiotherapy:
Radiation-induced myocardial fibrosis (RIMF) has both acute and chronic stages of RIHD, based on the specific pathology, and is thought to be a major risk factor for myocardial remodeling and vascular changes. Although patients may initially be asymptomatic, 10 years after RT, RIMF can subsequently result in restrictive or constrictive pericarditis and heart failure, which are lethal cardiovascular events.
“New therapeutic insights into radiation-induced myocardial fibrosis” (2019).
Stratton et al (2019) conducted an in vitro study to support the hypothesis that BRD4 controls activation of fibroblasts20:
Objective: We sought to test the hypothesis that BRD4 functions in a cell-autonomous and signal-responsive manner to control activation of cardiac fibroblasts, which are the major extracellular matrix (ECM)-producing cells of the heart.
Method and Results: RNA-sequencing (RNA-seq), mass spectrometry and cell-based assays employing primary adult rat ventricular fibroblasts (ARVFs) demonstrated that BRD4 functions as an effector of TGF-β signaling to stimulate conversion of quiescent cardiac fibroblasts into Periostin (Postn)-positive cells that express high levels of ECM.
Conclusions: These findings define BRD4 as a central regulator of the pro-fibrotic cardiac fibroblast phenotype, establish a p38-dependent signaling circuit for epigenetic reprogramming in HF, and uncover a novel role for Sertad4. The work provides a mechanistic foundation for the development of BRD4 inhibitors as targeted anti-fibrotic therapies for the heart.

BRD4 and arthritis
- Rheumatoid arthritis
Background to fibroblast-like synoviocytes (FLS):
Fibroblast-like synoviocytes (FLS) represent a specialised cell type located inside joints in the synovium. These cells play a crucial role in the pathogenesis of chronic inflammatory diseases, such as rheumatoid arthritis.
…The inner lining of the joint consists of the synovium (also called the synovial membrane), a thin layer located between the joint capsule and the joint cavity. The word "synovium" is derived from the word "synovia" (or synovial fluid), which is a clear, viscous fluid produced by the synovium, and its main purpose is to reduce friction between the joint cartilages during movement. Synovium is also important to maintain proper joint function by providing the structural support and supply of the necessary nutrients to the surrounding cartilage
…FLS that are present in the synovium during RA display altered phenotype compared to the cells present in normal tissues. They lose the property called contact inhibition (cells arrest their growth in the case when more cells come into contact with each other), and they also lose the growth dependency on adhesive surfaces; both these phenomena contribute to the increase in the number of FLS in the inflammatory tissue and are also typical for example for the growth of cancerous cells.
…The aggressive phenotype of FLS in RA and the effect these cells have on their microenvironment can be summarized into hallmarks that distinguish them from healthy FLS. These hallmark features of FLS in RA are divided into 7 cell-intrinsic hallmarks and 4 cell-extrinsic hallmarks. The cell-intrinsic hallmarks are: reduced apoptosis, impaired contact inhibition, increased migratory invasive potential, changed epigenetic landscape, temporal and spatial heterogeneity, genomic instability and mutations, and reprogrammed cellular metabolism. The cell-extrinsic hallmarks of FLS in RA are: promotes osteoclastogenesis and bone erosion, contributes to cartilage degradation, induces synovial angiogenesis, and recruits and stimulates immune cells.

From 2015, Zhang et al used a mouse model to perform an in vivo investigation into the effects of BRD4 inhibition on TNFa-stimulated human rheumatoid arthritis fibroblast-like synoviocytes (RAF-FLS) behaviour21.
RA is about more than just IL-16, IL-17 & Th17 cells, BRD4 is the missing link:
Highlights
BRD4 silencing reduced the proinflammatory cytokines from TNFα-stimulated RA-FLS.
Downregulation of BRD4 inhibited migration and invasion of RA-FLS.
BRD4 silencing decreased activation of c-Jun and NF-κB in TNFα-stimulated RA-FLS.
BRD4 inhibitor JQ1 reduced the inflammation and joint damage of CIA.
Abstract
We aimed to explore the effects of bromodomain-containing protein 4 (BRD4) inhibition on tumor necrosis factor (TNF)-α-stimulated human rheumatoid arthritis fibroblast-like synoviocytes (RA-FLS) behavior and the therapeutic implications using BRD4 inhibitor JQ1 were explored in vivo. The levels of interleukin (IL)-1β, IL-6, IL-17 and IL-18 in cultural supernatants from TNFα-stimulated RA-FLS were measured by ELISA. RA-FLS migration and invasion in vitro were investigated using wound healing and Matrigel assay. Expression of signaling pathway proteins was measured by Western blot. The in vivo effects of BRD4 inhibitor JQ1 were elucidated using collagen-induced arthritis (CIA) mice. We found BRD4 silencing reduced the secretion of IL-1β, IL-6, IL-17 and IL-18 from TNFα-stimulated human RA-FLS. Downregulation of BRD4 inhibited FBS-induced migration and invasion of human RA-FLS. BRD4 silencing decreased the phosphorylation of c-Jun and activation of NFκB in TNFα-stimulated RA-FLS. In vivo, BRD4 inhibitor JQ1 reduced the inflammatory response, autoantibody production and joint damage of CIA model. Our data suggest for the first time that BRD4 inhibition has anti-inflammatory property in RA.
The CIA model was used to explore the therapeutic implications of the BRD4 inhibitor JQ1 in vivo. As expected, treatment with JQ1 significantly reduced the incidence rate of CIA (90% vs 20%, P < 0.01) and led to a significant attenuation in the clinical arthritis severity score (Fig. 5A). Furthermore, histological examination was conducted to illustrate the joint pathology in CIA mice. Consistent with the clinical arthritis score, the JQ1-treated mice showed a significant reduction in joint inflammation and damage (Fig. 5B).
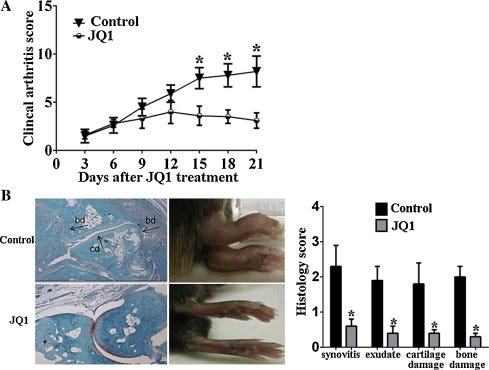
Over the years, cytokines have been demonstrated to be involved in RA pathology, and cytokine-blocking therapies have emerged from the hypothesis that the most abundant cytokines present in the joint were more likely to be pathogenic [22]. Numerous proinflammatory mediators including interleukin IL-1, IL-18, TNF and IL-17 are detected at the disease site, where they have a role in perpetuating inflammation, cartilage destruction, and bone remodelling associated with RA [23].
Synthetic mRNA gene agents also upregulate IL-1beta22:
IL-1β is a crucial mediator in the promoting synovial inflammation and pannus, as well as joint inflammation and bone destruction in RA [24], [25]. Importantly, IL-1β can induce expression of multiple proinflammatory mediators of innate immune reactions, and blockade of the IL-1 by the interleukin-1 receptor antagonist has demonstrated a central role of IL-1 in a number of human autoinflammatory diseases [26].
IL-17 induces the production of several inflammatory mediators in the diseased synovium, which are further synergistically enhanced via combinations of IL-17 with other cytokines, including IL-1β, IL-6, IL-17 and IL-18, contributing to the hyperplasia and exacerbated inflammation observed in joints of RA patients [30]. Furthermore, these IL-17-driven events initiate several feedback-loop mechanisms leading to increased expansion of Th17 cells and thereby increased production of IL-17 [31]. In this study, we showed that BRD4 inhibition markedly downregulated the expression of IL-1β, IL-6, IL-17, IL-18 and TNFα in vitro and in CIA mice. Additionally, histological analysis indicated that JQ1 treatment reduced the synovial inflammation and destruction in CIA mice.
Taken together, we present evidence for the first time that the BRD4 inhibition can inhibit joint inflammation and FBS-induced migration and invasion of human RA-FLS via decreasing the phosphorylation of c-Jun and activation of NFκB. In vivo, JQ1 treatments reduced the inflammatory response, autoantibody production and joint damage of CIA model. Our data suggest that BRD4 may be a new target for human RA.
- Acute gouty arthritis

Finally in this review, from 2020 and Hao et al evaluated the effects of the BRD4 inhibitor JQ-1 on acute gouty arthritis. They found that JQ-1 could effectively attenuate symptoms induced in rats23:
Results: Pretreatment of JQ1 and BRD4 siRNA significantly suppressed pyroptosis and inhibited activation of p65 NF-κB signaling as well as NLRP3 inflammasome in THP-1 cells exposed to MSU. In vivo, JQ-1 administration could effectively attenuate joint swelling and synovial inflammation in rats treated by intra-articular injection of MSU. More importantly, MSU led to macrophage pyroptosis and Brd4/NF-κB/NLRP3/GSDMD signaling induction in rat synoviums, which was improved by JQ-1.
Conclusions: Our study identifies the role of BRD4 in MSU-induced pyroptosis through regulating NF-κB/NLRP3/GSDMD signaling pathways, which provides a potential target for treatment of acute gouty arthritis.

BRD4 induced telomere lengthening of cancer cells
Not only does BRD4 induce a permanent SASP state in otherwise healthy cells and inhibits DNA repair, it can also help grant the gift of immortality to cancer cells through lengthening their telomeres. This also further helps inhibit the DDR.
Wang et al tested this in vitro in 2017 by using BET inhibitors to demonstrate a reversal of the effect24:
Cancer cells maintain telomere length equilibrium to avoid senescence and apoptosis induced by short telomeres, which trigger the DNA damage response. Limiting the potential for telomere maintenance in cancer cells has been long been proposed as a therapeutic target.
Using an unbiased shRNA screen targeting known kinases, we identified bromodomain-containing protein 4 (BRD4) as a telomere length regulator. Four independent BRD4 inhibitors blocked telomere elongation, in a dose-dependent manner, in mouse cells overexpressing telomerase.
Long-term treatment with BRD4 inhibitors caused telomere shortening in both mouse and human cells, suggesting BRD4 plays a role in telomere maintenance in vivo. Telomerase enzymatic activity was not directly affected by BRD4 inhibition. BRD4 is in clinical trials for a number of cancers, but its effects on telomere maintenance have not been previously investigated.
DMSO is the negative control, “KU” or KU55933 is the positive control and JQ1 is the BET inhibitor:

BRD4 translocations are linked to cancer, and BRD4 inhibition reduces cell proliferation in acute myeloid leukemia (63,64), potentially by blocking BRD4 mediated transcription of c-MYC. Our finding, that BRD4 inhibition shortens telomeres in human and mouse cells in culture, suggests that part of the mechanism by which BRD4 inhibitors block cancer cell growth (64–66) may be through telomere shortening.
BRD4 inhibitors are currently in early phase clinical trials for treatment of hematopoietic cancers, including acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), multiple myeloma and lymphoma (67). If telomere shortening also occurs in vivo in a clinical trial setting, telomere shortening might accentuate the anti-cancer effects of BRD4. BRD4 also affects immune cell function through its interaction with NF-κB (68), and BRD4 inhibitors have been proposed as anti-inflammatory agents, including in liver fibrosis and idiopathic pulmonary fibrosis (69,70). However, these approaches should be carefully considered, because short telomeres in humans lead to telomere syndromes that can manifest as bone marrow failure, pulmonary fibrosis, liver fibrosis and other diseases (71–73).
Telomere shortening in either cancer clinical trials or fibrosis may exacerbate underlying short telomere syndromes. Stratifying patients by telomere length before treatment may help identify individuals who might be at risk from side effects caused by further telomere shortening.
BRD4 and diabetes
There are several papers investigating the link between BRD4 and diabetes.
For instance, Nord et al (2022) conducted an in vitro study into how BET inhibitors can disrupt BRD4-p65 interactions and reduce inducible nitric oxide synthase transcription in pancreatic β-cells25.
They used rat insulinoma cell line INS 832/13 as these provide a useful model for studying insulin secretion regulation and pancreatic islet beta-cell function26:
Chronic inflammation of pancreatic islets is a key driver of β-cell damage that can lead to autoreactivity and the eventual onset of autoimmune diabetes (T1D). In the islet, elevated levels of proinflammatory cytokines induce the transcription of the inducible nitric oxide synthase (iNOS) gene, NOS2, ultimately resulting in increased nitric oxide (NO). Excessive or prolonged exposure to NO causes β-cell dysfunction and failure associated with defects in mitochondrial respiration. Recent studies showed that inhibition of the bromodomain and extraterminal domain (BET) family of proteins, a druggable class of epigenetic reader proteins, prevents the onset and progression of T1D in the non-obese diabetic mouse model. We hypothesized that BET proteins co-activate transcription of cytokine-induced inflammatory gene targets in β-cells and that selective, chemotherapeutic inhibition of BET bromodomains could reduce such transcription.
Here, we investigated the ability of BET bromodomain small molecule inhibitors to reduce the β-cell response to the proinflammatory cytokine interleukin 1 beta (IL-1β). BET bromodomain inhibition attenuated IL-1β-induced transcription of the inflammatory mediator NOS2 and consequent iNOS protein and NO production. Reduced NOS2 transcription is consistent with inhibition of NF-κB facilitated by disrupting the interaction of a single BET family member, BRD4, with the NF-κB subunit, p65. Using recently reported selective inhibitors of the first and second BET bromodomains, inhibition of only the first bromodomain was necessary to reduce the interaction of BRD4 with p65 in β-cells. Moreover, inhibition of the first bromodomain was sufficient to mitigate IL-1β-driven decreases in mitochondrial oxygen consumption rates and β-cell viability. By identifying a role for the interaction between BRD4 and p65 in controlling the response of β-cells to proinflammatory cytokines, we provide mechanistic information on how BET bromodomain inhibition can decrease inflammation. These studies also support the potential therapeutic application of more selective BET bromodomain inhibitors in attenuating β-cell inflammation.
This further suggests that the ability of BET inhibitors to disrupt NF-κB signaling is enacted at the point of transcriptional activation in the nucleus, rather than upstream signaling cascade components in the cytoplasm. These findings are consistent with BRD4 co-activating transcription of the NF-κB target gene NOS2 through a bromodomain-dependent interaction with p65 in the nucleus of β-cells.
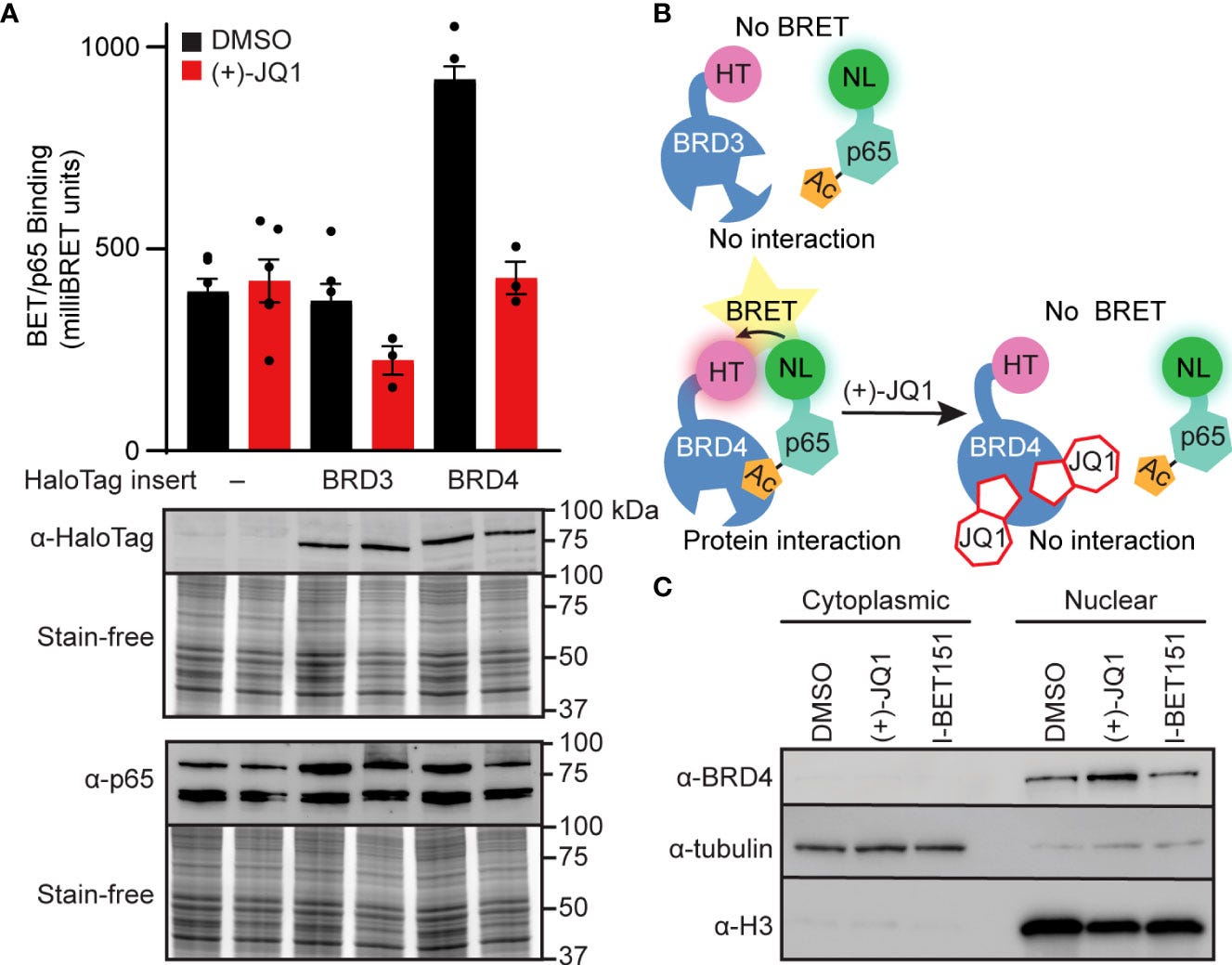
FIGURE 3 BET bromodomain inhibitors disrupt BRD4-p65 interaction. (A) NanoBRET assays completed in INS 832/13 cells transfected with Halo-tagged BET tandem bromodomains and nanoluciferase-tagged p65 exposed to 1 μM (+)-JQ1 or DMSO before BRET intensity reading. Expression blots show relative amounts of transfected proteins. (B) Schematic interpretation of NanoBRET results. (C) Immunoblot with an antibody directed against BRD4 in INS 832/13 cell fractions after 4-hour exposure to 0.5 μM (+)-JQ1, 1 μM I-BET151, or DMSO. Error bars for nanoBRET assay represent the standard error of the mean (SEM) from 3-6 individually plated wells per treatment condition.https://www.frontiersin.org/files/Articles/923925/fendo-13-923925-HTML-r1/image_m/fendo-13-923925-g003.jpg
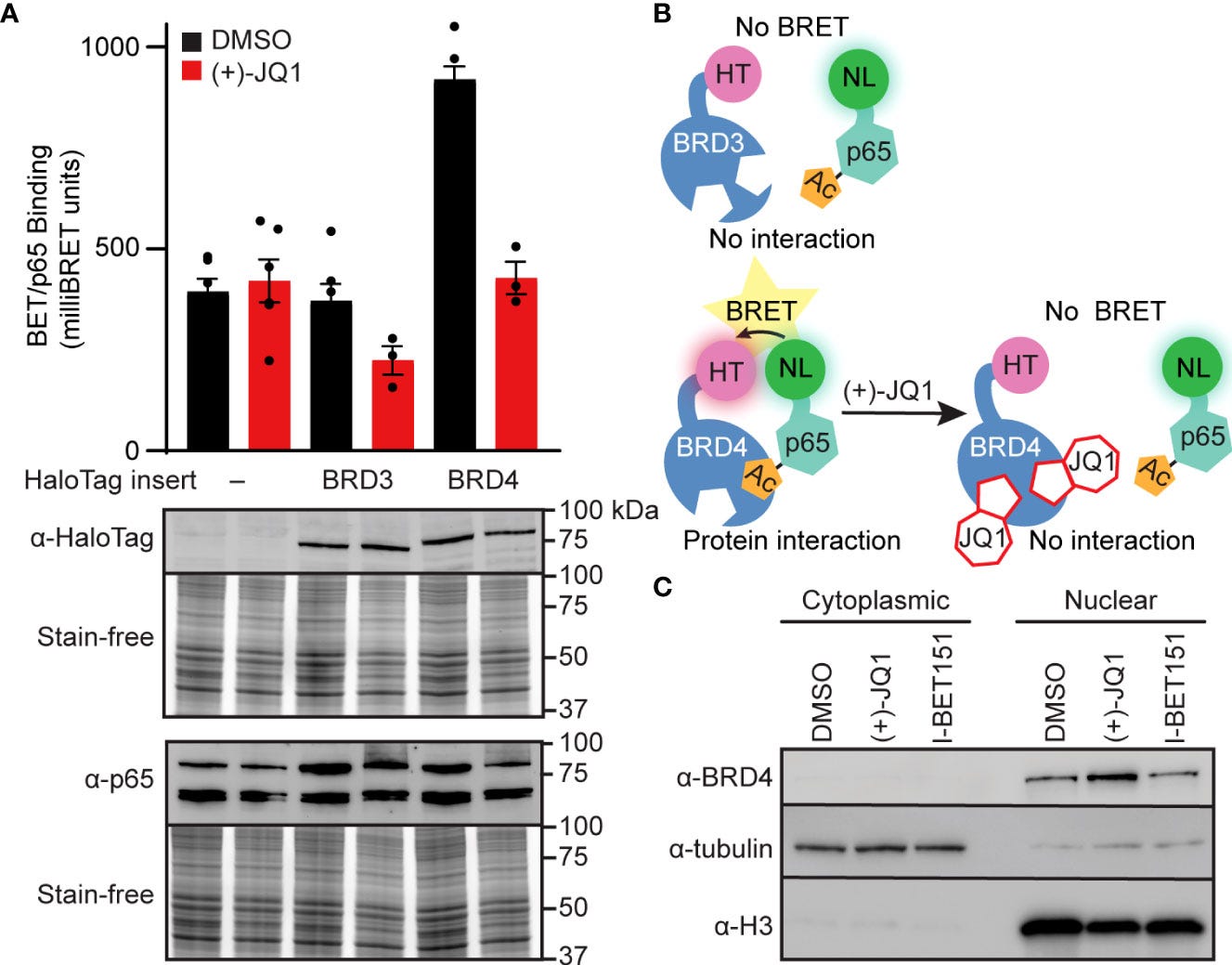
{kind=link}
{kind=link}
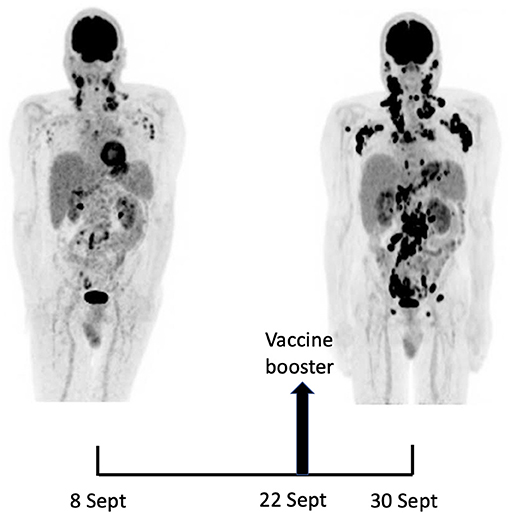{kind=link}
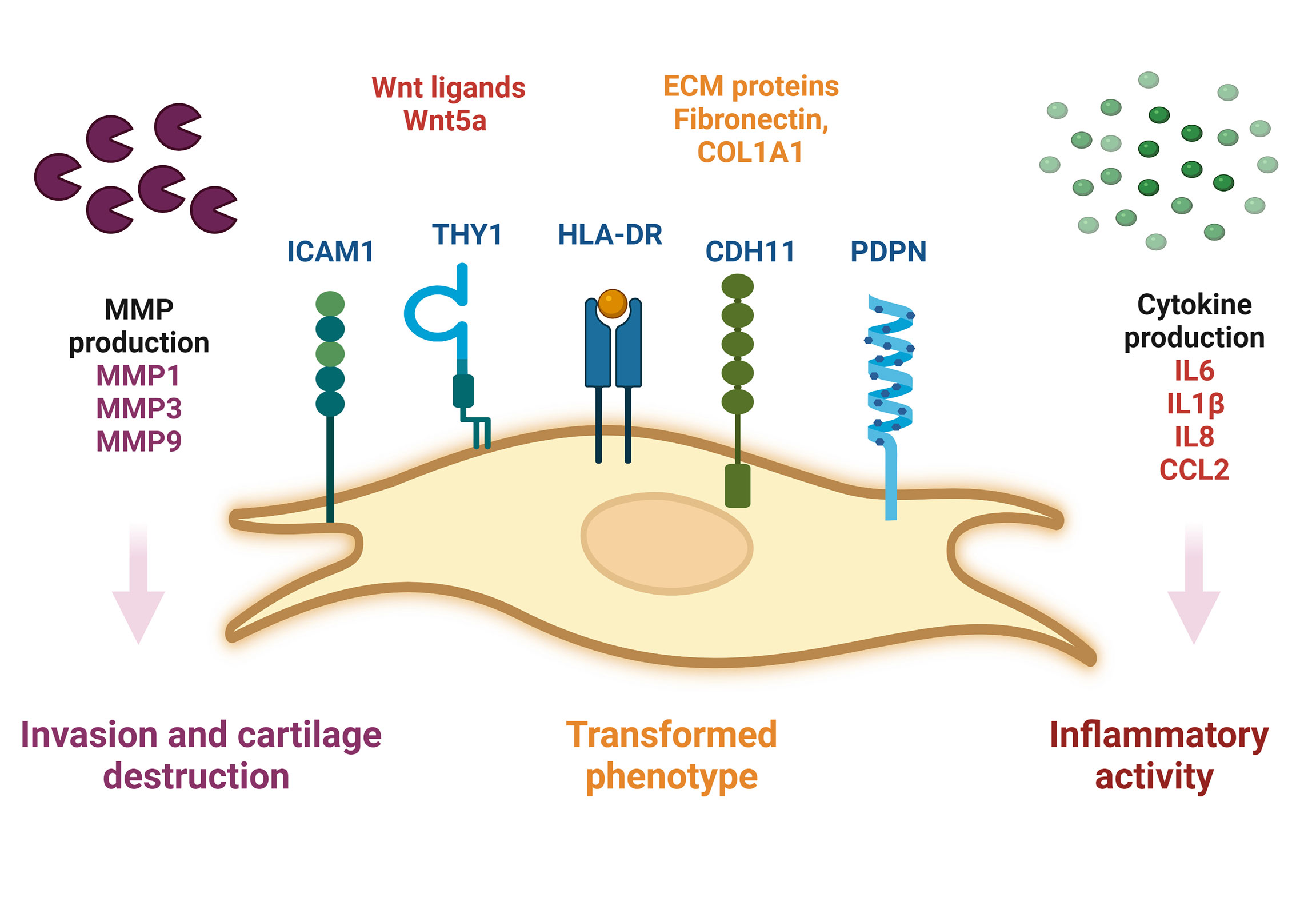{kind=link}
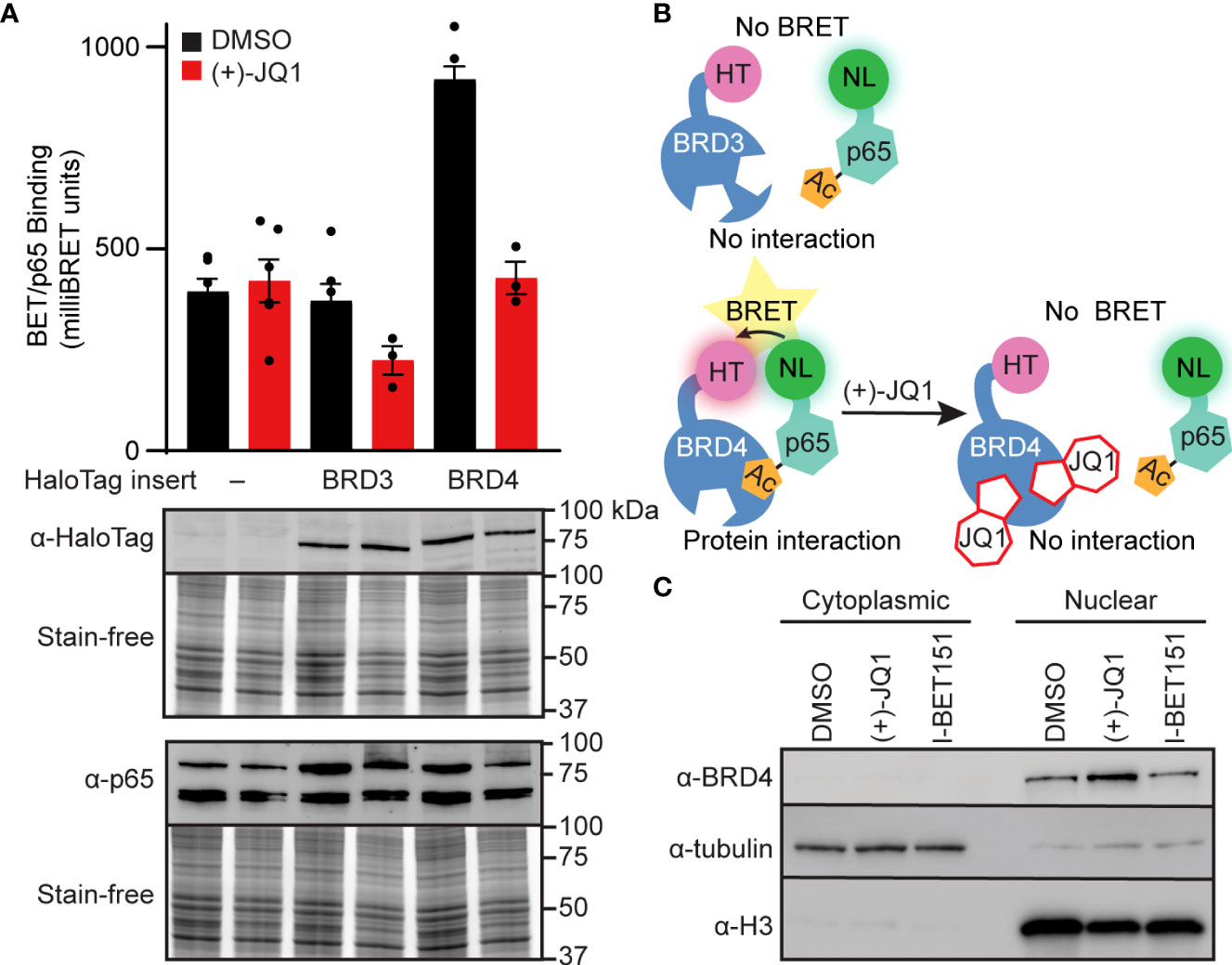{kind=link}
And finally, in 2022 Lipinski et al submitted a paper at the American Chemical Society Fall 2022 conference: “Dual glucosamine-BET bromodomain inhibitor acts cooperatively to inhibit transcription of NF-κB target genes in pancreatic β-cells”27. The full version is paywalled.
In brief, they hypothesise that a glucosamine analogue of the BRD4 inhibitor JQ1 would be more effective at treating T1D, and with less off-target side effects, than using JQ1 alone, ie synergistic effects. So far, they have conducted in silico modelling and it has been tested on rat islet models of T1D:
Abstract
Islets of Langerhans are a heterogeneous mixture of endocrine (hormone-secreting) and exocrine cells in the mammalian pancreas. Pancreatic β-cells are the most abundant islet cell-type and maintain glucose homeostasis by secreting insulin in response to increased blood glucose concentrations. Type 1 diabetes (T1D), for which there is no cure, is characterized by the autoimmune destruction of β-cells. The bromodomain and extra-terminal domain (BET) family of epigenetic reader proteins have emerged as novel drug targets to prevent T1D. However, current BET bromodomain inhibitors (BETi) are associated with adverse, off-target effects in clinical trials primarily for cancer indications. Any cure for T1D would be widely used in a pediatric patient population so avoiding off-target effects is imperative. We propose to ameliorate negative effects of BET bromodomain inhibition by targeting BETi to pancreatic β-cells. Here, we exploited the high abundance of glucose transporters on the β-cell surface relative to other cells in the pancreas and conjugated a BETi to glucosamine (GlcN). BETi attenuate gene transcription induced by the pro-inflammatory cytokine IL-1β in the NF-κB signaling. Moreover, GlcN also inhibits gene production in the NF-κB pathway; thus, we hypothesized that our dual BETi-GlcN would have increased efficacy as an anti-inflammatory agent. We synthesized a novel, GlcN-based analogue of JQ1, [JQ1-GlcN], in which the BETi JQ1 was conjugated to GlcN. Molecular modeling and biophysical assays showed that JQ1-GlcN retains binding to both bromodomains of BRD4 in vitro. JQ1-GlcN is selectively active in cell-based and primary rat islet models of T1D. In summary, we developed a novel chemical probe and pharmacological agent selective for pancreatic endocrine cells to harness the full potential of BETi both to mechanistically study the roles of BET bromodomains in the pathophysiology of T1D and selectively target BET proteins in β-cells as a potential therapy to prevent or treat T1D.
I would hypothesise that if you took quercetin, for example, with glucosamine sulphate then you may achieve similar synergies. But please do not change any meds without consulting a qualified healthcare professional first.
On checking PubMed for this combination it was effective when combined with chondroitin to treat osteoarthritis, but not rheumatoid arthritis:
In this study, forty-six OA and twenty-two RA patients were administered with the GCQG supplement orally for 3 months. Several parameters of the knee joints were monitored before and after supplementation. The OA patients showed a significant improvement in pain symptoms, daily activities (walking and climbing up and down stairs), and visual analogue scale, and changes in the synovial fluid properties with respect to the protein concentration, molecular size of hyaluronic acid, and chondroitin 6-sulphate concentration were also observed. However, no such effects were observed in the RA patients. These results suggest that the GCQG supplement exerted a special effect on improving the synovial fluid properties in OA patients.
From: “Effects of an oral administration of glucosamine-chondroitin-quercetin glucoside on the synovial fluid properties in patients with osteoarthritis and rheumatoid arthritis” (2009).
Quercetin alone as a treatment for T2D.
Comparisons to metformin and the potential correction of insulin resistance is of particular note here:
Herein we investigated the molecular mechanism of action of the citrus flavonoid, quercetin in skeletal muscle cells (L6 myotubes). Taking advantage of protein kinase inhibitors, we proved that the effect of quercetin on 2-NBDG uptake in L6 myotubes was not through insulin signaling pathway, but through adenosine monophosphate kinase (AMPK) pathway and its downstream target p38 MAPK. An increase in the cellular AMP to ATP ratio on pretreatment may account for AMPK activation which was coupled with a transient change in mitochondrial membrane potential. In addition, quercetin triggered a rise in intracellular calcium suggesting that calcium-calmodulin mediated protein kinase (CaMKK) may also be involved. Quercetin shared a similar mechanism with the well-known drug metformin, highlighting it as a promising compound for the management of type 2 diabetes. The AMPK signaling pathway could contribute to correction of insulin resistance through bypassing the insulin-regulated system for GLUT4 translocation.
From: “Quercetin, a Lead Compound against Type 2 Diabetes Ameliorates Glucose Uptake via AMPK Pathway in Skeletal Muscle Cell Line” (2017)

Disclaimer
This site is strictly an information website about potential therapeutic agents and a review of associated research papers. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
References
Mekawy AS, Alaswad Z, Ibrahim AA, Mohamed AA, AlOkda A, Elserafy M. The consequences of viral infection on host DNA damage response: a focus on SARS-CoVs. J Genet Eng Biotechnol. 2022;20:104. doi:10.1186/s43141-022-00388-3
Meyer K, Patra T, Vijayamahantesh, Ray R. SARS-CoV-2 Spike Protein Induces Paracrine Senescence and Leukocyte Adhesion in Endothelial Cells. J Virol. 95(17):e00794-21. doi:10.1128/JVI.00794-21
Heo JY, Seo YB, Kim EJ, et al. COVID-19 vaccine type-dependent differences in immunogenicity and inflammatory response: BNT162b2 and ChAdOx1 nCoV-19. Frontiers in Immunology. 2022;13. Accessed July 18, 2023. https://www.frontiersin.org/articles/10.3389/fimmu.2022.975363
Zhang P, Li R, Xiao H, et al. BRD4 Inhibitor AZD5153 Suppresses the Proliferation of Colorectal Cancer Cells and Sensitizes the Anticancer Effect of PARP Inhibitor. Int J Biol Sci. 2019;15(9):1942-1954. doi:10.7150/ijbs.34162
Tang M, Hu X, Wang Y, et al. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacological Research. 2021;163:105207. doi:10.1016/j.phrs.2020.105207
https://www.sciencedirect.com/science/article/pii/S1043661820315152
‘We Made a Big Mistake’ — COVID Vaccine Spike Protein Travels From Injection Site, Can Cause Organ Damage. Children’s Health Defense. Accessed July 21, 2023. https://childrenshealthdefense.org/defender/covid-vaccine-spike-protein-travels-from-injection-site-organ-damage/
Pfizer report_Japanese government.pdf. Accessed July 21, 2023. https://www.docdroid.net/xq0Z8B0/pfizer-report-japanese-government-pdf
Biodistribution of Pfizer Covid-19 Vaccine | Regenerative Medicine Center - Dr. Valerie Donaldson MD Pittsburgh, Pennsylvania PA. Accessed July 21, 2023. https://regenerativemc.com/biodistribution-of-pfizer-covid-19-vaccine/
Sheets RL, Stein J, Bailer RT, et al. Biodistribution and Toxicological Safety of Adenovirus Type 5 and Type 35 Vectored Vaccines Against Human Immunodeficiency Virus-1 (HIV-1), Ebola, or Marburg Are Similar Despite Differing Adenovirus Serotype Vector, Manufacturer’s Construct, or Gene Inserts. J Immunotoxicol. 2008;5(3):315-335. doi:10.1080/15376510802312464
Zhang Y, Peng X, Xue M, et al. SARS-COV-2 spike protein promotes RPE cell senescence via the ROS/P53/P21 pathway. Biogerontology. Published online February 4, 2023:1-15. doi:10.1007/s10522-023-10019-0
Takahashi K, Yi H, Liu CH, et al. Spinal bromodomain-containing protein 4 contributes to neuropathic pain induced by HIV glycoprotein 120 with morphine in rats. NeuroReport. 2018;29(6):441. doi:10.1097/WNR.0000000000000992
Zhu X, Enomoto K, Zhao L, et al. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clinical Cancer Research. 2017;23(2):430-440. doi:10.1158/1078-0432.CCR-16-0914
Floyd SR, Pacold ME, Huang Q, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498(7453):246-250. doi:10.1038/nature12147
https://europepmc.org/backend/ptpmcrender.fcgi?accid=PMC3683358&blobtype=pdf
Targeting ACE2-BRD4 in Cancer and COVID-19 | Cancer Community (nature.com)
TSUKAMOTO T, NAKAHATA S, SATO R, et al. BRD4-Regulated Molecular Targets in Mantle Cell Lymphoma: Insights into Targeted Therapeutic Approach. Cancer Genomics Proteomics. 2020;17(1):77-89. doi:10.21873/cgp.20169
Dang Q, Sun Z, Wang Y, Wang L, Liu Z, Han X. Ferroptosis: a double-edged sword mediating immune tolerance of cancer. Cell Death Dis. 2022;13(11):1-16. doi:10.1038/s41419-022-05384-6
Schmitt A, Grimm M, Kreienkamp N, et al. BRD4 inhibition sensitizes diffuse large B-cell lymphoma cells to ferroptosis. Blood. Published online June 9, 2023:blood.2022019274. doi:10.1182/blood.2022019274
Goldman S, Bron D, Tousseyn T, et al. Rapid Progression of Angioimmunoblastic T Cell Lymphoma Following BNT162b2 mRNA Vaccine Booster Shot: A Case Report. Frontiers in Medicine. 2021;8. Accessed July 20, 2023. https://www.frontiersin.org/articles/10.3389/fmed.2021.798095
https://www.frontiersin.org/articles/10.3389/fmed.2021.798095/full
Martella N, Pensabene D, Varone M, et al. Bromodomain and Extra-Terminal Proteins in Brain Physiology and Pathology: BET-ing on Epigenetic Regulation. Biomedicines. 2023;11(3):750. doi:10.3390/biomedicines11030750
Stratton MS, Bagchi RA, Felisbino MB, et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ Res. 2019;125(7):662-677. doi:10.1161/CIRCRESAHA.119.315125
Zhang Q gang, Qian J, Zhu Y chang. Targeting bromodomain-containing protein 4 (BRD4) benefits rheumatoid arthritis. Immunol Lett. 2015;166(2):103-108. doi:10.1016/j.imlet.2015.05.016
https://www.sciencedirect.com/science/article/abs/pii/S0165247815000929?via%3Dihub
Adenovirus-based but not mRNA-based vaccines transiently alter platelet count homeostasis in healthy subjects. ISTH Congress Abstracts. Accessed July 19, 2023. https://abstracts.isth.org/abstract/adenovirus-based-but-not-mrna-based-vaccines-transiently-alter-platelet-count-homeostasis-in-healthy-subjects/
Hao K, Jiang W, Zhou M, et al. Targeting BRD4 prevents acute gouty arthritis by regulating pyroptosis. Int J Biol Sci. 2020;16(16):3163-3173. doi:10.7150/ijbs.46153
Wang S, Pike AM, Lee SS, Strong MA, Connelly CJ, Greider CW. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017;45(14):8403-8410. doi:10.1093/nar/gkx561
Nord JA, Wynia-Smith SL, Gehant AL, et al. N-terminal BET bromodomain inhibitors disrupt a BRD4-p65 interaction and reduce inducible nitric oxide synthase transcription in pancreatic β-cells. Front Endocrinol (Lausanne). 2022;13:923925. doi:10.3389/fendo.2022.923925
https://www.frontiersin.org/articles/10.3389/fendo.2022.923925/full
INS-1 832/13 Rat Insulinoma Cell Line | SCC207. Accessed July 20, 2023. https://www.merckmillipore.com/GB/en/product/INS-1-832-13-Rat-Insulinoma-Cell-Line,MM_NF-SCC207
Dual glucosamine-BET bromodomain inhibitor acts cooperatively to inhibit transcription of NF-κB target genes in pancreatic β-cells. Published August 21, 2022. Accessed July 20, 2023. https://www.morressier.com/o/event/62daeef3a6fd3a00196fa00a/article/630fcb4d7e215f5e7f37454e
Thanky ou again for the time and effort you put into these literature reviews. They are much appreciated. However you find me puzzled by one paragraph:
"He could discuss this and argue that despite being associated with tumorigenesis the agent stays in the arm, isn’t there for long enough etc, but he doesn’t. Its now a binary “yes” or “no” argument without consideration of underlying cancer pathways, just a lot of arm waving and SHOUTING."
You are aware of the biodistribution study #185350 which Pfizer handed over to the FDA end of 2020, right? Cause in that study it is shown that 15 minutes after IM injection the LNPs can be detected in ever tissue and every organ of the body... to be honest I would be hugely surprised if you had not seen that study before. Then again, since you can mostly find it on the www.phmpt.org page, and the FDA has introduced its new misdirection campaign (apart from misinformation, disinformation and malinformation) "Rumor-Control", it might be that people will disregard it.
Link to stduy #185350 https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M4_4223_185350.pdf (table 1, pages23-24)
And my take on the FDA's attempt to paint themselves as the "beacon of truth in the sea of lies":
https://ghostfromthefuture.substack.com/p/fact-checking-is-so-yesterday-rumor (Yes, a judge told them to 'produce' the facts, but forgot to tell them to do so on their official page; so what is the best way to hide the evidence anyways? Make sure that people don't trust their eyes when they see it, or as the Georg Orwell said: "The Party told you to reject the evidence of your eyes and ears. It was their final, most essential command.")