


Megathread #33: SARS-CoV-2 infectivity of CD4+ & CD26+/DPP4 T-lymphocyte cells, Tat-like motifs, CCR5, integrin binding and related pathologies

Reading time: about 75 minutes.
Also available in other languages:
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
If received via email I recommend clicking on the hyperlinked title to read the latest version in full, in a browser.

Contents
Simian immunodeficiency virus (SIV)
HIV, GP120 and binding to DC-SIGN & CD4
RANTES (CCL5) & chemokine receptor 5 (CCR5)
Integrins and the arginine-glycine-aspartic acid (RGD) sequence
Other studies into antiplatelet autoimmunity
Introduction
As is the case with many of us, I was through with COVID pathologies and wanted to move on to writing about other health related matters, but it turned out that spike protein related pathologies aren’t quite through with me yet: more keep presenting themselves in the literature.
Indeed in the last month I discovered papers relating to 4 more cancer related pathways, the last one concerning bromodomain-4:
The fourth will review spike protein binding to protein phosphatase 2 (PP2).
But before we get to that, this Substack concerns spike protein binding to CD4+ T cells, and the mechanisms involved with this extend far beyond the induction of lymphopenia.
As per convention I will define key terminology as we go. If you keep these in mind it helps give you a much better handle on understanding the pathologies involved, but I will keep it as brief as possible.
CD: The cluster of differentiation (also known as cluster of designation or classification determinant and often abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules providing targets for immunophenotyping of cells. In terms of physiology, CD molecules can act in numerous ways, often acting as receptors or ligands important to the cell. A signal cascade is usually initiated, altering the behavior of the cell (see cell signaling). Some CD proteins do not play a role in cell signaling, but have other functions, such as cell adhesion. CD for humans is numbered up to 371 (as of 21 April 2016.)
If you are deficient in CD4+ T helper cells then your CD8 killer cells are also rendered ineffective by not being signalled into action:
CD4: In molecular biology, CD4 (cluster of differentiation 4) is a glycoprotein that serves as a co-receptor for the T-cell receptor (TCR). CD4 is found on the surface of immune cells such as T helper cells, monocytes, macrophages, and dendritic cells. It was discovered in the late 1970s and was originally known as leu-3 and T4 (after the OKT4 monoclonal antibody that reacted with it) before being named CD4 in 1984. In humans, the CD4 protein is encoded by the CD4 gene.
CD4+ T helper cells are white blood cells that are an essential part of the human immune system. They are often referred to as CD4 cells, T-helper cells or T4 cells. They are called helper cells because one of their main roles is to send signals to other types of immune cells, including CD8 killer cells, which then destroy the infectious particle. If CD4 cells become depleted, for example in untreated HIV infection, or following immune suppression prior to a transplant, the body is left vulnerable to a wide range of infections that it would otherwise have been able to fight.
T lymphocytopenia was, until recently, usually associated with HIV:
Lymphocytopenia is the condition of having an abnormally low level of lymphocytes in the blood. Lymphocytes are a white blood cell with important functions in the immune system. It is also called lymphopenia. The opposite is lymphocytosis, which refers to an excessive level of lymphocytes.
Lymphocytopenia may be present as part of a pancytopenia, when the total numbers of all types of blood cells are reduced.
In T lymphocytopenia, there are too few T lymphocytes, but normal numbers of other lymphocytes. It causes, and manifests as, a T cell deficiency. This is usually caused by HIV infection (resulting in AIDS), but may be Idiopathic CD4+ lymphocytopenia (ICL), which is a very rare heterogeneous disorder defined by CD4+ T-cell counts below 300 cells/μL in the absence of any known immune deficiency condition, such as human immunodeficiency virus (HIV) infection or chemotherapy.
On 8th July ‘23 I posted the following link, which I researched further this week in reply Dr Kevin McCairn’s link to the same study (hat tip to @Maples46014332):

This study was posted 11th January ‘23 by Brunetti et al, and it has a very long list of authors in comparison to most papers1.
Its a pre-print, but that no longer means what it used to mean in regards to the relative value, merits or reproducibility of a study when compared to fully peer reviewed papers.
The best advice I can give is to try to understand the signalling pathways involved as best you can, read around the subject as much as possible, don’t always rely just on the wording of the abstract and most importantly see what other research there is out there which came to similar conclusions.
Conflict of interest (COI) statements rarely disclose much but you can see what other papers the authors have pin their biographies against ResearchGate, for example:

One of the objectives of my reviews is to do as much as the leg work as possible for you and I will always add new findings as amendments if needed, good or bad. My limitations are that I do not have the time or the resources to do meta-analysis of thousands of papers but most of the time you don’t need to, it can muddy the waters and you can cherry pick whatever conclusions you like out of a big enough orchard, so there are advantages and disadvantages - my aim is to present the evidence which quite often is scant in number or hidden away on journal sites or even retracted if it doesn’t fit the political (or shareholder focused) narrative of the day.
Brunetti et al conducted multiple studies and confirmed that CD4+ T helper cells can be directly infected by SARS-CoV-2, as evidenced by in silico, in vitro and ex vivo confirmation of RNA in cells taken from COVID-19 patients:
“SARS-CoV-2 Uses CD4 to Infect T Helper Lymphocytes” (2023):
From the abstract:
The mechanism by which SARS-CoV-2 infection may result in immune system dysfunction is still not fully understood. Here we show that SARS-CoV-2 infects human CD4+ T helper cells, but not CD8+ T cells, and is present in blood and bronchoalveolar lavage T helper cells of severe COVID-19 patients.
Interleukin 10 is an anti-inflammatory, immune suppressing cytokine as the body attempts to prevent a cytokine storm via negative feedback2:
We demonstrated that SARS-CoV-2 spike glycoprotein (S) directly binds to the CD4 molecule, which in turn mediates the entry of SARS-CoV-2 in T helper cells. This leads to impaired CD4 T cell function and may cause cell death. SARS-CoV-2-infected T helper cells express higher levels of IL-10, which is associated with viral persistence and disease severity. Thus, CD4-mediated SARS-CoV-2 infection of T helper cells may contribute to a poor immune response in COVID-19 patients.
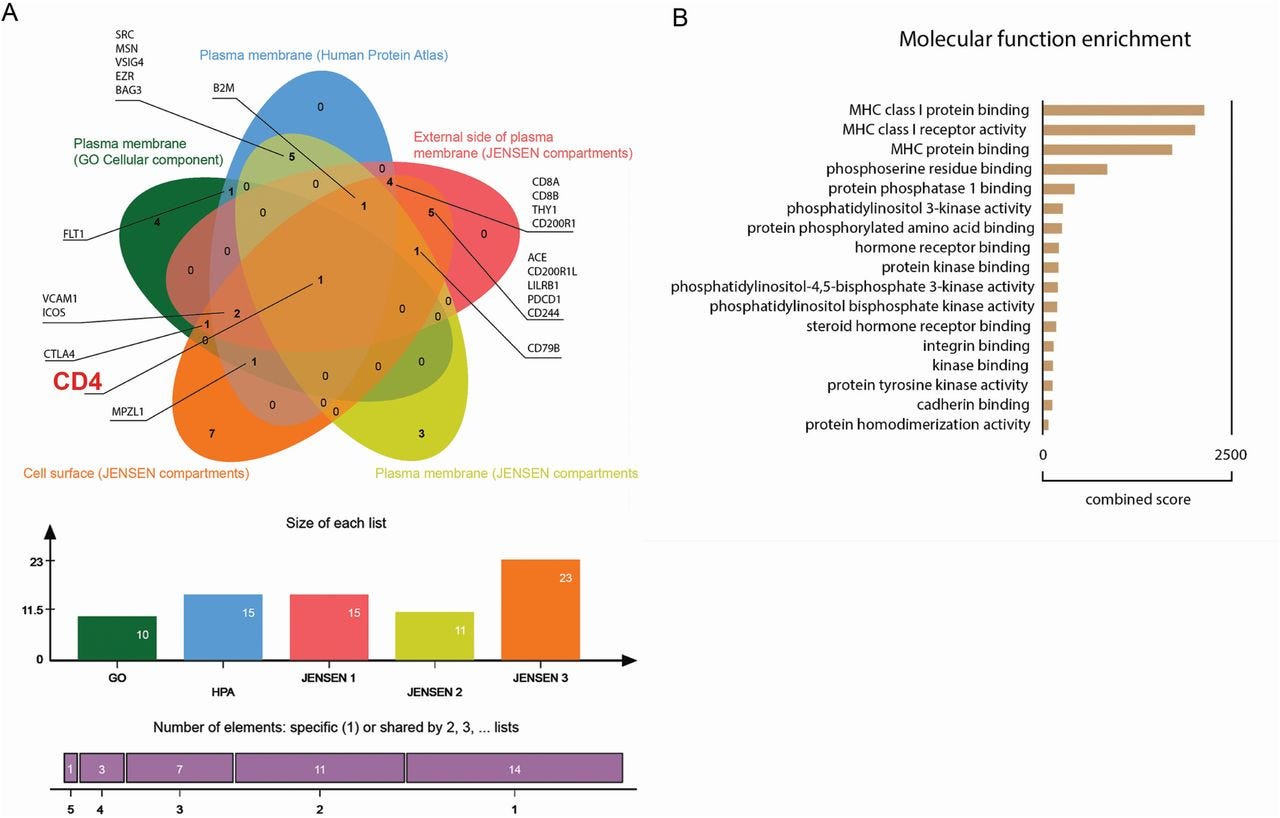
Since the structures of the spike of SARS-CoV-1 (sCoV-1) and the sCoV-2 proteins are similar, we used the P-HIPSTer algorithm to uncover human proteins that putatively interact with spike. Seventy-one human proteins were predicted to interact with sCoV-1 (Extended Data Fig. 3). We then cross-referenced the proteins with five databases of plasma membrane proteins to identify the ones located on the cell surface (see Methods for details). CD4 was the only protein predicted to interact with sCoV-1 that appeared in all five databases (Extended Data Fig.3). CD4 is expressed mainly in T helper lymphocytes and has been shown to be the co-receptor to HIV24. Since CD4+ T lymphocytes orchestrate innate and adaptive immune response, infection of CD4+ T cells by SARS-CoV-2 could explain lymphocytopenia and dysregulated inflammatory response in severe COVID-19 patients. Moreover, from an evolutionary perspective, infection of CD4+ T cells represents an effective mechanism for viruses to escape the immune response.

Extended Data Fig. 3 (A) Venn diagram of human proteins that were predicted by P-HIPSTer to interact with sCoV-1 and considered as part of cellular membrane in at least one database. Proteins found in at least two databases are annotated. (B) Molecular function enrichment of 71 predicted human proteins that interact with sCoV-1 according to P-HIPSTer. https://www.medrxiv.org/content/10.1101/2020.09.25.20200329v2.full To investigate whether SARS-CoV-2 infects CD4+ T cells in vivo, we purified CD4+ and CD8+ T cells from peripheral blood cells of COVID-19 patients (Table S1). Similar to our ex vivo experiments, SARS-CoV-2 RNA was detected in CD4+ T cells from COVID-19 patients (Fig. 1F). Using publicly available single-cell RNA sequencing data, we detected the presence of SARS-CoV-2 RNA in 2.1% of CD4+ T cells of the bronchoalveolar lavage (BAL) of patients with severe COVID-19 (Fig. 1G and Extended Data Fig. 5). Thus, our data demonstrate that SARS-CoV-2 infects CD4+ T cells.

Figure 1. SARS-CoV-2 infects CD4+ T cells in vitro and in vivo Peripheral blood CD3+CD4+ and CD3+CD8+ were infected with mock control or SARS-CoV-2 (CoV-2) (MOI 1) for 1 h under continuous agitation. (A) Viral load was assessed by RT-qPCR 24 h after infection. (B) Cells were washed, cultured for 24 h, fixed with PFA 4% and stained with RdRp probe for in situ hybridization or anti-sCoV-2 for immunofluorescence. Cells were analyzed by confocal microscopy. (C) C1-C5 - Representative transmission electron microscopy (EM) micrographs showing viral particles (asterisks) inside lymphocytes 2 h after infection. (D) Viral load was determined by qPCR 2 h, 6 h, 12 h, 24 h and 48 h after infection. (E) Vero cells were incubated with the supernatant of mock control or CoV-2 infected CD4+ T cells under continuous agitation for 1 h. The viral load in Vero cells was measured after 72 h using plaque assay. PFU – plaque-forming unit. (F) Viral load was measured by RT-qPCR in peripheral blood CD4+ and CD8+ T cells from healthy controls (HC) and COVID-19 patients. (G) CoV-2 RNA detection in CD4+ T cells from bronchoalveolar lavage (BAL) single-cell sequencing data revealing the presence of CoV-2 RNA. Data represents mean ± SEM of at least two independent experiments performed in triplicate or duplicate (f). ***p < 0.001 compared to all. ND=not detected. https://www.medrxiv.org/content/10.1101/2020.09.25.20200329v2.full

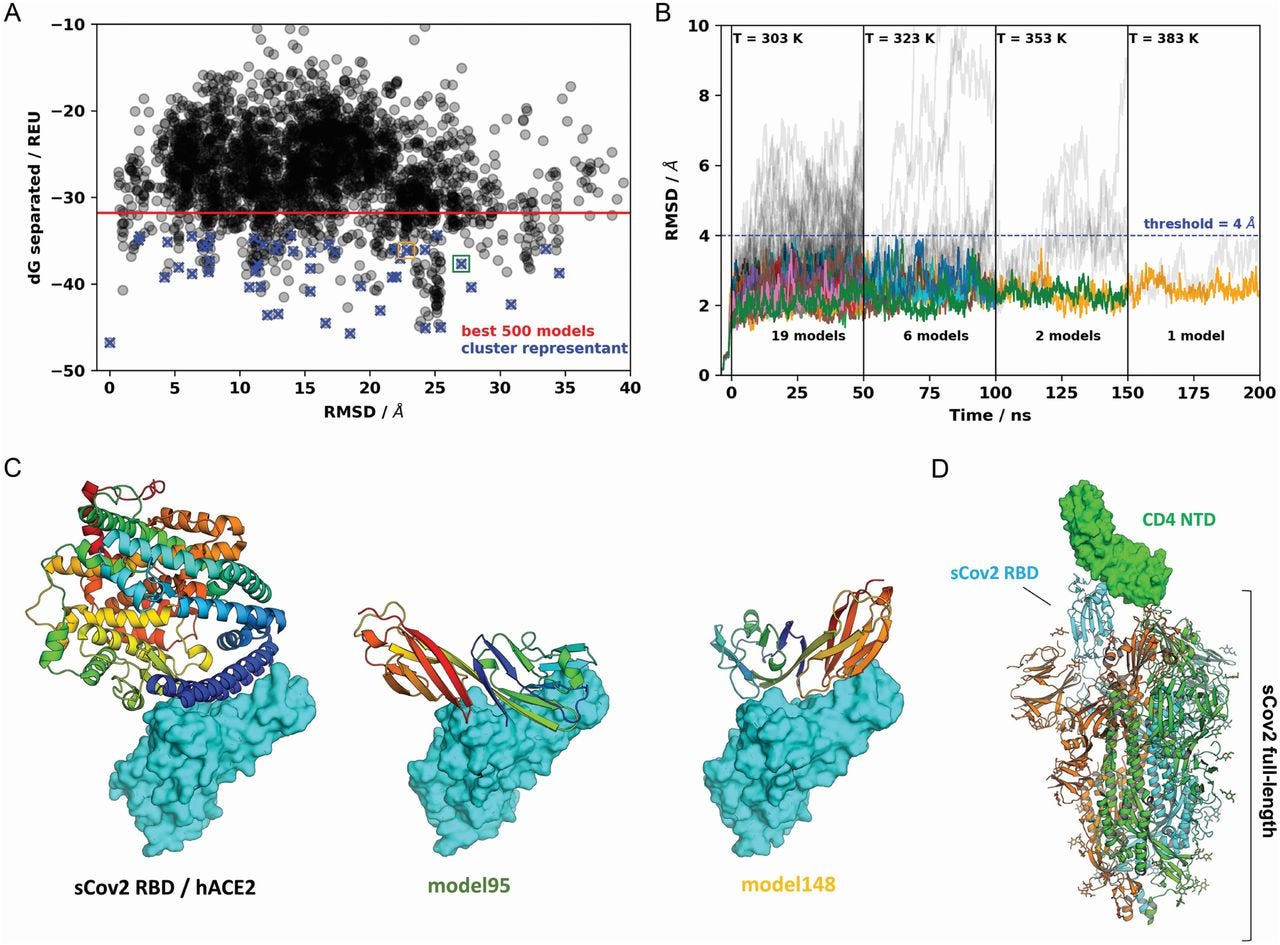
Our immunoprecipitation experiments indicated no physical interaction between CD4 and ACE2 (data not shown). Since CD4+ T cells have very low ACE2 expression, we tested whether CD4 alone was sufficient to allow SARS-CoV-2 entry. Inhibition of ACE2 using polyclonal antibody (Fig. 3I) diminished SARS-CoV-2 entry in CD4+ T cells. Moreover, the inhibition of TMPRSS2 with camostat mesylate reduced SARS-CoV-2 load (Fig. 3H). Altogether, these data demonstrate that ACE2, TMPRSS2 and CD4 are all required to allow the infection of CD4+ T cells by SARS-CoV-2.
Note that in the whole paper there is no mention of mRNA gene transfection and it is encouraging to see a focus on “complementary therapeutic approaches” instead.
Long lasting or cumulative effects are a major concern, especially if you keep getting reinfected or boosted:
How long these alterations in T cell function persist in vivo and whether they have long-lasting impacts on adaptive immunity remains to be determined. Hence, avoiding T cell infection by blocking sCoV-2-CD4 interaction and boosting T cell resistance against SARS-CoV-2 might represent complementary therapeutic approaches to preserve immune response integrity and prevent patients from progressing to the severe stages of COVID-19.
Up until now I had associated COVID-19 or spike protein binding and fusion with CD4+’s to be due to glycoprotein 120 binding as per the 2020 paper by Pradhan et al3.
From the abstract:
We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag.
Key point here, even though the full 120 Daltons (molecular weight) of gp120 peptide are missing they don’t necessarily need to be present. The pattern of molecular charge over the presenting amino acids in the motif and protein folding & structure are more important for determining binding affinities to the target receptor than specific sequences: think more of the RBD motifs as being like a key and the rest of the spike protein as the handle, in simple terms.

Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature.
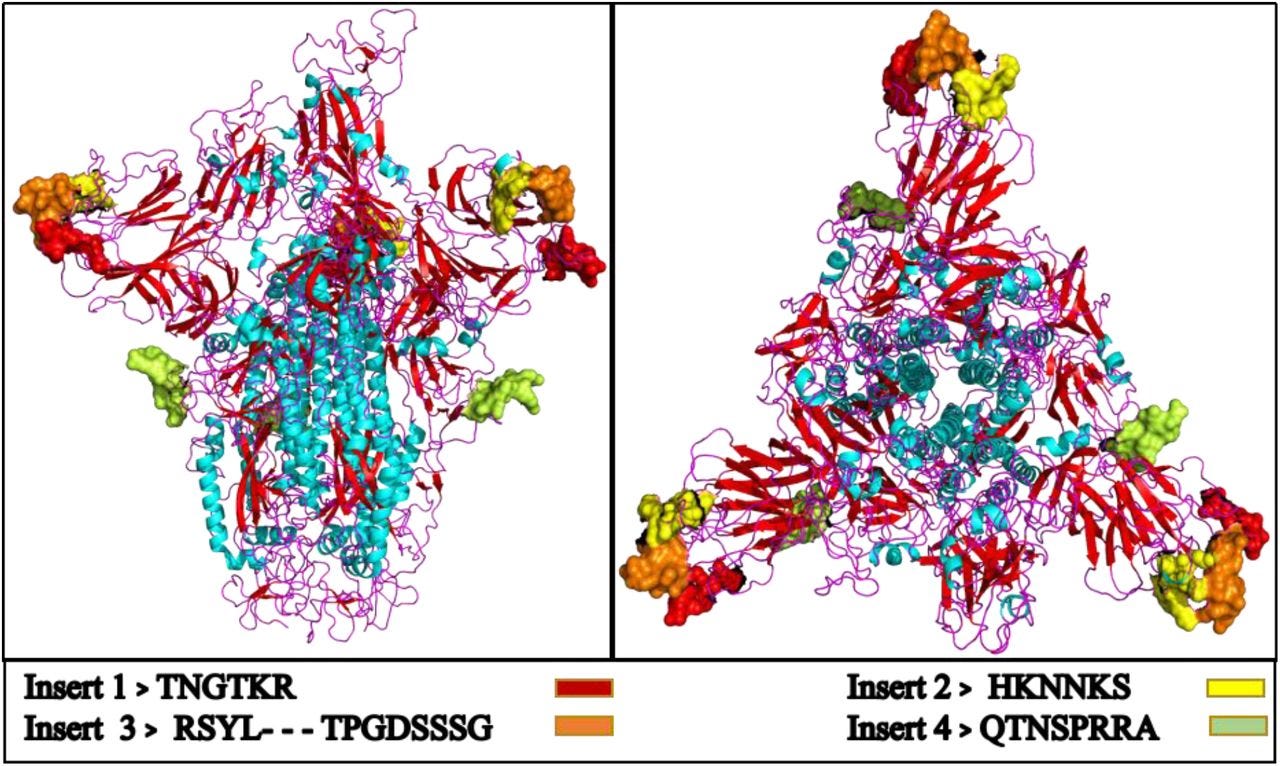
The amino acid residues of inserts 1, 2 and 3 of 2019-nCoV spike glycoprotein that mapped to HIV-1 were a part of the V4, V5 and V1 domains respectively in gp120 [Table 1]. Since the 2019-nCoV inserts mapped to variable regions of HIV-1, they were not ubiquitous in HIV-1 gp120, but were limited to selected sequences of HIV-1 [refer S.File1] primarily from Asia and Africa.
The HIV-1 Gag protein enables interaction of virus with negatively charged host surface (Murakami, 2008) and a high positive charge on the Gag protein is a key feature for the host-virus interaction.
In addition the spike glycoproteins are critical targets for vaccine development (Du et al., 2013). For this reason, the spike proteins represent the most extensively studied among coronaviruses. We therefore sought to investigate the spike glycoprotein of the 2019-nCoV to understand its evolution, novel features sequence and structural features using computational tools.
I make no apologies for reposting this. Apart from through breakthrough infections the risk hasn’t gone away. They are still pushing this stuff mostly to a cohort of the population least able to withstand the panopoly of pathophysiologies to follow (or the coercion involved), as per megathread posts #1-32 in this series and a host of case reports and papers.
They know that these mRNA gene agents do not prevent infection or transmission and are associated with AE’s worse than the disease, aegrescit medendo, and will be based on the XBB.1.5 variant, which will be long extinct by then4. In fact it was already in 30% weekly decline in prevalence by 20th July, only a month after they locked down production (yellow shading)5🤡:

“The full list of patients to be offered a Covid-19 booster jab by the NHS6”:
residents in a care home for older adults
all adults aged 65 years and over
persons aged from 6 months to 64 years who are in a clinical risk group
frontline health and social care workers
persons aged 12 to 64 years who are household contacts of people with immunosuppression
persons aged 16 to 64 years who are carers and staff working in care homes for older adults
Simian immunodeficiency virus (SIV)
As for HIV, there are various timelines but it appears to be a novel virus that made its first appearance in the latter half of the twentieth century, coincident with research into tumour inducing viruses and its wild origins in SIV, which for millennia was not readily transmissible to or replication competent in humans until something changed coincident with said research. The repeated weaponization of gp120 appears to be one of the outcomes.
Wikipedia goes all “Scooby Couey” on us to explain this coincidence, they don’t reference any gain of function research:
Simian immunodeficiency virus (SIV) is a species of retrovirus that cause persistent infections in at least 45 species of non-human primates. Based on analysis of strains found in four species of monkeys from Bioko Island, which was isolated from the mainland by rising sea levels about 11,000 years ago, it has been concluded that SIV has been present in monkeys and apes for at least 32,000 years, and probably much longer.
Virus strains from three of these primate species, SIVsmm in sooty mangabeys, SIVgor in gorillas and SIVcpz in chimpanzees, are believed to have crossed the species barrier into humans, resulting in HIV-2 and HIV-1 respectively, the two HIV viruses. The most likely route of transmission of HIV-1 to humans involves contact with the blood of chimps and gorillas that are often hunted for bushmeat in Africa. Four subtypes of HIV-1 (M, N, O, and P) likely arose through four separate transmissions of SIV to humans, and the resulting HIV-1 group M strain most commonly infects people worldwide. Therefore, it is theorized that SIV may have previously crossed the species barrier into human hosts multiple times throughout history, but it was not until recently, after the advent of modern transportation and global commuterism, that it finally took hold, spreading beyond localized decimations of a few individuals or single small tribal populations.

From the 24th December 1971 issue of Science, and the somewhat secretive researchers in Special Virus Cancer Program or SVCP even then were held in low esteem by their peers.
The SVCP’s goals were regarded more as a political rather than scientific, and lacking in evidence of intellectual underpinning.
Launched in 1964, it used military and space program (ie military) planning techniques, was not open to criticism, was said to give too much power to individual scientists and appeared to be more interested in isolating cancer causing viruses rather than gaining “an understanding of the fundamental aspects of cancer and cell biology“.
I will leave it to the reader as to what the real goal was of the SVCP, and does this all sound familiar given the developments of last three years?

HIV, GP120 and binding to DC-SIGN & CD4
As for HIV, GP120 and CD4+ T cells the route of infection appears to be circuitous rather than by direct infection:
DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin) also known as CD209 (Cluster of Differentiation 209) is a protein which in humans is encoded by the CD209 gene.[5]
DC-SIGN is a C-type lectin receptor present on the surface of both macrophages and dendritic cells.
Role in HIV infection
This molecule is involved in the initial stages of the human immunodeficiency virus infection, as the HIV gp120 molecule causes co-internalization of the DC-SIGN molecule and HIV virus particle (virion). The dendritic cell then migrates to the cognate lymphoid organ, whereupon recycling of the DC-SIGN/HIV virion complex to the cell periphery facilitates HIV infection of CD4+ T cells by interaction between DC-SIGN and ICAM-3.
A paper from 1995 by Dianzani et al discusses the conundrum in “Modulation of CD4 lateral interaction with lymphocyte surface molecules induced by HIV-1 gp120”7.
From the abstract:
CD4, a lymphocyte surface glycoprotein, serves as co-receptor for antigen with the T cell receptor (TCR). It is also the lymphocyte receptor for HIV by binding the gp120 viral envelope protein. Interaction of gp120 with CD4 is crucial for viral infection, but is not sufficient to allow viral entry into cells. Recombinant gp120 alters CD4+ T cell responsiveness to activation stimuli. To express its co-receptor function fully, CD4 must be laterally associated with the TCR and CD45 to form multi-receptor complexes competent to transduce potent activation signals.
In the absence of gp120, CD4 displayed high association with CD3, CD5, CD45RC, CD25, CD28, CD44, and CD53; weak association with CD2, CD38, CD45RB, CD62L, and CD26; and no association with CD45RA, CD45RO, CD11b, CD11a, CD54, CD7, CD48, CD98, CD59, CD55, HLA class I and class II molecules.
Treatment with gp120 significantly increased CD4 association with CD3, CD45RA, CD45RB, CD59, CD38, CD26, and HLA class I, and decreased that with CD45RC.
RANTES (CCL5) & chemokine receptor 5 (CCR5)
RANTES (CCL5) is interesting as it is also helps mediate HIV infection of CD4+ T-cells by binding to CCR5:
Chemokine (C-C motif) ligand 5 (also CCL5) is a protein which in humans is encoded by the CCL5 gene. The gene has been discovered in 1990 by in situ hybridisation and it is localised on 17q11.2-q12 chromosome. It is also known as RANTES (regulated on activation, normal T cell expressed and secreted). RANTES was first described by Dr. Tom Schall who named the protein, the original source of the name Rantes was from the Argentine movie Man Facing Southeast about an alien who shows up in a mental ward who was named Rantés, the rather clunky acronym was only made to fit the name.
…The chemokine CCL5 is mainly expressed by T-cells and monocytes, and it has not been shown to be expressed by B-cells. Moreover, it is abundantly expressed by epithelial cells, fibroblasts and thrombocytes. Although it can bind to receptors CCR1, CCR3, CCR4 and CCR5, belonging to seven transmembrane G-protein coupled receptor (GPCRs) family, it has the highest affinity to the CCR5.
…In cytotoxic T-cells (CTL) killing other cells via Fas/FasL interaction, CCL5 increases HIV-specific T-cell cytotoxicity. Moreover, it is considered that CCL5 in low concentration might inhibit HIV replication. It binds to CCR5 (as well as 2 other chemokines) on the surface of CD4+ T-cells. CCR5 is used by HIV as an entrance molecule to a cell. On the contrary, CCL5 in high concentration might increase HIV replication.
 - IMDb")
If you follow the pathology for long enough the same names seem to keep cropping up. For example, a genotyping study from 2021 by Hubacek et al associated a 32 nucleotide deletion from the CCR5 gene with protection from both HIV and SARS-CoV-2 infection.
They must both be using the same receptor, another coincidence.
“CCR5Δ32 Deletion as a Protective Factor in Czech First-Wave COVID-19 Subjects”8.
From the abstract:
Significant inter-individual differences have been observed during the course of the infection, which suggests that genetic susceptibility may be a contributing factor. CC chemokine receptor 5 (CCR5), which acts as a co-receptor for the entry of HIV-1 into cells, is promising candidate whose can have an influence on SARS-CoV-2 infection.
A genetic mutation known as CCR5Δ32, consisting of a 32-nucleotide deletion, encodes a truncated protein that protects homozygous carriers of the deletion from HIV-1 infection. Similarly, inhibition of CCR5 seems to be protective against COVID-19. In our study, we successfully genotyped 416 first-wave SARS-CoV-2-positive infection survivors (164 asymptomatic and 252 symptomatic) for CCR5Δ32, comparing them with a population based sample of 2,404 subjects.
We found the highest number (P=0.03) of CCR5Δ32 carriers in SARS-CoV-2-positive/COVID-19-asympto-matic subjects (23.8 %) and the lowest number in SARS-CoV-2-positive/COVID-19-symptomatic patients (16.7 %), with frequency in the control population in the middle (21.0 %). We conclude that the CCR5Δ32 I/D polymorphism may have the potential to predict the severity of SARS-CoV-2 infection.
Another smoking gun, until recently the CCR5Δ32 mutation was only specifically associated with demonstrable protection from the HIV virus.
Key extracts from “C-C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection” by Barmania and Pepper (2013):
In 1996, it was discovered that CCR5 is necessary as a co-receptor for entry of the macrophage tropic HIV strains (Dragic et al., 1996, Deng et al., 1996). Dragic et al. demonstrated that the ligands for CCR5 inhibit viral entry and envelope mediated fusion (Dragic et al., 1996). The virus uses CCR5 especially during initial infection, whereas the alternative co-receptor ‘C-X-C chemokine receptor type four’ (CXCR4) is used much later in HIV infection when the infected individual is progressing towards AIDS.
In individuals infected with HIV, the percentage of CD4+CCR5+ T-cells is higher (13.2%) than when compared to uninfected individuals (6.2%) (Ostrowski et al., 1998). The highest percentage of expression was found in an individual with acute HIV syndrome, recorded at around 30–40%. The variation in CCR5 percentages in HIV infected individuals did not correlate with genotype as three individuals with heterozygosity for Δ32, a CCR5 mutation known to reduce HIV infection (reviewed in Section 7), had different levels of expression (2.7%, 13.1% and 17%). In contrast, the activation state of the CD4+ cells as measured by HLA-DR positively correlated with CCR5 expression.
In 1999, a study conducted by de Roda Husman et al. assessed CCR5 expression in terms of CCR5 genotype and HIV infection and progression (de Roda Husman et al., 1999)…The study postulated that the CD4+CCR5+ T-cell percentage is directly correlated with HIV disease progression due to the constant immune activation associated with HIV. The presence of the CCR5 receptor on memory effector T-cells (Mo et al., 1998) or mature activated T-cells supports the latter finding.
Martinson et al. analyzed the distribution of the Δ32 mutation in more than 3000 individuals from various countries and found a 2–5% gene frequency in Europe, Middle East and some parts of the Indian subcontinent (Martinson et al., 1997). Isolated incidences of Δ32 found in other regions were attributed to European gene flows into these areas. The highest frequency of the mutation was discovered in the Ashkenazi Jewish population at frequencies of 20.93%. In 2005, Novembre et al. confirmed these results when they assessed the Δ32 frequency in various population groups worldwide (Novembre et al., 2005). The mutant allele is absent in Black populations excluding the African American group which may have acquired the mutation through admixture.
The origin of the Δ32 mutant allele was dated back to between 275 and 1875 years ago, as a unique mutation that increased over the years due to a selective pressure (Stephens et al., 1998). Stephans et al. used haplotype analysis on the chromosomes of 192 Caucasian individuals and estimated the origin using a coalescence theory (Stephens et al., 1998). They hypothesized that the Black plague was the strong selective pressure that caused the mutant allele boom.
Historical data however suggests that the Black plague is not the selective force. The distribution of Δ32 in a north to south gradient does not correspond to casualties of the plague. In fact, the distribution follows a south to north gradient (Cohen and Weaver, 2006). Moreover, the Black plague showed the greatest casualties and effects in areas with the lowest allele frequencies of Δ32, such as the Mediterranean region and China.
Smallpox was another pandemic deemed culpable for the Δ32 mutant allele increase (Galvani and Slatkin, 2003). The pandemic had severe casualties that exceeded those of the plague. Smallpox is a virus similar to HIV, as poxviruses are known to infect lymphocytes using chemokine receptors (Novembre et al., 2005). Conversely, historical evidence refutes this theory as smallpox started outside Europe and did not affect anyone country more significantly than another (Cohen and Weaver, 2006). The discovery of ancient DNA with similar allele frequencies of Δ32 indicated that historic pandemics such as the plague and smallpox did not result in the allele increase.
The hype surrounding the Δ32 mutation stems from its ability to protect homozygous individuals from HIV. However, in 1997, studies showed that some individuals with homozygous Δ32 were infected with HIV (Biti et al., 1997, O'Brien et al., 1997, Theodorou et al., 1997). Analysis of the HIV strains in these individuals revealed the presence of X4 utilizing HIV, accompanied by very rapid CD4+ T-cell decline (Michael et al., 1998). This indicates that the mutation does not protect Δ32 homozygous individuals from viral strains which utilize alternative receptors.
The protective effect of the Δ32 mutation towards HIV is a consequence of an inhibition of CCR5 protein expression on the cell surface. This prevents HIV from utilizing the receptor for viral entry. In addition, the Δ32 protein, localized to the endoplasmic reticulum, exerts a trans-dominant negative effect on the wild type CCR5 protein, inhibiting its transport to the cell surface (Benkirane et al., 1997, Chelli and Alizon, 2001).
CD26 (DPP4) & Tat protein
I was aware of the involvement of HIV with CD26 (DPP4 or DPPIV) from other studies too, which prompted me to follow it up further:
Dipeptidyl peptidase-4 (DPP4), also known as adenosine deaminase complexing protein 2 or CD26 (cluster of differentiation 26) is a protein that, in humans, is encoded by the DPP4 gene.
DPP-4 is known to cleave a broad range of substrates including growth factors, chemokines, neuropeptides, and vasoactive peptides. The cleaved substrates lose their biological activity in the majority of cases, but in the case of the chemokine RANTES and neuropeptide Y, DPP-4 mediated cleavage leads to a shift in the receptor subtype binding.

From 1994 and Morimoto et al conducted an in vitro examination into the “Role of CD26/dipeptidyl peptidase IV in human immunodeficiency virus type 1 infection and apoptosis9”.
The main text is a pdf, key highlights:
ABSTRACT To examine the role of CD26/dipeptidyl peptidase IV (DPPIV; EC 3.4.14.5) in infection by human immunodeficiency virus type 1 (HIV-1), we utilized CD26 cDNA-transfected Jurkat T-cell lines. Both CD26- parental Jurkat cells and mutant CD26+ (DPPIV-) transfected Jurkat cells were readily infected with HIV-1, whereas wild-type CD26+ (DPPIV+) transfected Jurkat cells were more resistant to HIV-1 infection. Our results suggest that CD26 is not essential for HIV-1 infectivity as suggested by others but that DPPIV enzyme activity may decrease the efficiency of HIV-1 infection. Of great interest, we found that mutant CD26+ (DPPIV-) transfectants and CD26- parental Jurkat cells strongly expressed CD95 (Fas/Apo-1) and were more sensitive than wild-type CD26+ (DPPIV+) transfectants to the induction of apoptosis by anti-CD95 monoclonal antibody. These results suggest that CD26 may play a role in HIV-1-associated loss of CD4+ cells through the process of programmed cell death.
CD4+ lymphocytes in patients with acquired immunodeficiency syndrome (AIDS) have an intrinsic defect in their ability to recognize and respond to "recall antigens" some time before a reduction in the total number of CD4+ cells occurs (11, 12). The response to recall antigens is clearly a property of CD4+CD26+ T cells, since this is the only helper population known to proliferate in response to soluble antigens and to induce both major histocompatibility complex-restricted cytotoxic T lymphocytes, capable of killing virally infected target cells and B cells to secrete immunoglobulins.
A reduction in CD26+ T cells may occur prior to a more generalised loss of other CD4+T cells. However this would not be detected by typical WBC counts.
Each of these properties is a key to the host's response to viral infection. In this regard, a selective decrease in CD26+ T cells has been reported in human immunodeficiency virus type 1 (HIV-1)-infected individuals prior to a general decrease in CD4+ T cells (13, 14).
The role of Tat protein in infection by HIV:
In molecular biology, Tat is a protein that is encoded for by the tat gene in HIV-1. Tat is a regulatory protein that drastically enhances the efficiency of viral transcription. Tat stands for "Trans-Activator of Transcription".
Tat also appears to play a more direct role in the HIV disease process. The protein is released by infected cells in culture, and is found in the blood of HIV-1 infected patients.
It can be absorbed by cells that are not infected with HIV, and can act directly as a toxin producing cell death via apoptosis in uninfected "bystander" T cells, assisting in progression toward AIDS.
By antagonizing the CXCR4 receptor, Tat also appears to selectively encourage the reproduction of less virulent M-tropic (macrophage-tropic) strains of HIV (which use the CCR5 receptor) early in the course of infection, allowing the more rapidly pathogenic T-tropic (T-cell-tropic) strains (which use the CXCR4 receptor) to emerge later after mutating from M-tropic strains

Moreover, Tat, a regulatory protein encoded by the HIV-1 genome (15), has been shown to suppress the response of human peripheral T cells to soluble antigens (16). It has been suggested that Tat can bind to and partially inhibit DPPIV (17). More recently, Hovanessian and colleagues (18) have proposed that CD26 serves as an essential cofactor for HIV-1 entry, capable of rendering CD4-expressing murine NIH 3T3 cells infectible by HIV-1. These authors found that the anti-CD26 mAb 1F7 and inhibitors of DPPIV enzyme activity could inhibit viral entry.
We can probably let gp120 off the hook this time:
In addition, we found no evidence to support the view that gp120 either binds to or is a substrate for recombinant soluble CD26, arguing strongly against the suggestion that DPPIV has endopeptidase activity capable of cleaving the V3 loop of gp120 during HIV-1 entry. Moreover, it is clear from Table 1 that parental CD26- Jurkat cells can be infected with HIV-1 despite a lack of CD26 antigen expression or measurable DPPIV enzymatic activity. Others have shown that CD26 is not required for syncytium formation and some have been unable to confim Hovanessian and coworkers' studies (18) on viral entry (30-32). Taken together, these data suggest that CD26 is not a necessary cofactor for HIV infection.
In conclusion, you may well have a Tat protein induced selective loss of CD4+ T helper cells before you become symptomatic:
We suggest that the HIV Tat protein can contribute to both diminished immune responsiveness and enhanced susceptibility to the induction of apoptosis through its binding to CD26. The HIV-1 Tat protein is known to be essential for transactivation of viral genes as well as for viral replication (15) and can also be detected in the sera of HIV patients (16, 17). HIV-1 Tat has also been shown to suppress the response of human peripheral T cells to recall antigen in vitro (16, 17), a property of the CD4+CD26+ T cell. More importantly, HIV-1 Tat protein can bind to CD26 and partially inhibit DPPIV enzyme activity (16). As shown in Fig. 3, we found not only that the CD95 molecule was strongly expressed on mutant CD26+ (DPPIV-) T-cell lines but that these CD26+ (DPPIV-) enzyme-deficient lines were highly susceptible to both HIV-1 infection and the induction of apoptosis. Our in vitro observations suggest an important role for CD26 in HIV infectivity and provide a clinically relevant mechanism to partially explain both the selective loss of CD4+ helper cells and helper cell function in asymptomatic HIV-infected patients.
“Crystal Structures of HIV-1 Tat-derived Nonapeptides Tat-(1–9) and Trp2-Tat-(1–9) Bound to the Active Site of Dipeptidyl-peptidase IV (CD26)” (2005)
DPPIV has been shown to play a crucial role in T-cell activation and in several functions of the immune system (2, 36, 37, 38). Previous evidence implicated DPPIV in antigen-specific T-cell activation events and Tat protein in suppression of antigen-induced, but not mitogen-induced, T-cell proliferation. In the present work, we show that the N-terminal residues of Tat-(1–9) bind to the active site of DPPIV and provide evidence that Tat-(1–9) might inhibit DPPIV activity. This suggests that the immunosuppressive effect of Tat is mediated, at least partly, by competitive inhibition of DPPIV activity. In addition, the inhibition of DPPIV activity influences the processing of chemokines SDF-1α, SDF-1β, RANTES, and LD78β, thereby modulating their anti-HIV activities.

https://www.sciencedirect.com/science/article/pii/S0021925820660148?via%3Dihub
Integrins and the arginine-glycine-aspartic acid (RGD) sequence
And yet going back to previous studies we know that SARS-CoV-2 can infect CD4+ T cells directly. But if its not through gp120 then does it have a Tat like motif? This would be yet another binding mechanism in addition to via other receptors or by direct binding to CD4.
An in vitro investigation from 1999 by Barillari et al quickly signposted what to look for in SARS-CoV-2: “The Tat Protein of Human Immunodeficiency Virus Type-1 Promotes Vascular Cell Growth and Locomotion by Engaging the α5β1 and αvβ3 Integrins and by Mobilizing Sequestered Basic Fibroblast Growth Factor”10.
First mention of Tat in relation to other cells, cancer and fibrosis too, as well as binding to integrins. You really must try to limit your exposure to Tat or its motifs:
Key extracts:
The Tat protein of human immunodeficiency virus type-1 (HIV-1) has been shown to be released during acute infection of T cells by HIV-1 and to promote angiogenesis and Kaposi’s sarcoma (KS) development in infected individuals. In this study, we investigated the molecular mechanisms responsible for the angiogenic effects of Tat.
The results shown herein indicate that two different Tat domains cooperate to induce these effects by different pathways. The arginine-glycine-aspartic acid (RGD) sequence present at the carboxyterminal of Tat mediates vascular cell migration and invasion by binding to the α5β1 and αvβ3 integrins.
The C-terminus (also known as the carboxyl-terminus, carboxy-terminus, C-terminal tail, C-terminal end, or COOH-terminus) is the end of an amino acid chain (protein or polypeptide), terminated by a free carboxyl group (-COOH).
This interaction also provides endothelial cells with the adhesion signal they require to grow in response to mitogens. At the same time, the Tat basic sequence retrieves into a soluble form extracellular basic fibroblast growth factor (bFGF) bound to heparan sulfate proteoglycans by competing for heparin-binding sites. This soluble bFGF mediates Tat-induced vascular cell growth. These effects resemble those of extracellular matrix proteins, suggesting that Tat enhances angiogenesis and promotes KS progression by a molecular mimicry of these molecules.
Integrins are transmembrane receptors that help cell-cell and cell-extracellular matrix (ECM) adhesion. Upon ligand binding, integrins activate signal transduction pathways that mediate cellular signals such as regulation of the cell cycle, organization of the intracellular cytoskeleton, and movement of new receptors to the cell membrane. The presence of integrins allows rapid and flexible responses to events at the cell surface (e.g. signal platelets to initiate an interaction with coagulation factors).
Several types of integrins exist, and one cell generally has multiple different types on its surface. Integrins are found in all animals while integrin-like receptors are found in plant cells.
Integrins work alongside other proteins such as cadherins, the immunoglobulin superfamily cell adhesion molecules, selectins and syndecans, to mediate cell–cell and cell–matrix interaction. Ligands for integrins include fibronectin, vitronectin, collagen and laminin.
…They are also involved in a wide range of other biological activities, including extravasation, cell-to-cell adhesion, cell migration, and as receptors for certain viruses, such as adenovirus, echovirus, hantavirus, and foot-and-mouth disease, polio virus and other viruses. Recently, the importance of integrins in the progress of autoimmune disorders is also gaining attention of the scientists. These mechanoreceptors seem to regulate autoimmunity by dictating various intracellular pathways to control immune cell adhesion to endothelial cell layers followed by their trans-migration. This process might or might not be dependent on the sheer force faced by the extracellular parts of different integrins.
…Integrins play an important role in cell signaling by modulating the cell signaling pathways of transmembrane protein kinases such as receptor tyrosine kinases (RTK).
…Knowledge of the relationship between integrins and receptor tyrosine kinase has laid a foundation for new approaches to cancer therapy. Specifically, targeting integrins associated with RTKs is an emerging approach for inhibiting angiogenesis.
…Integrins have an important function in neuroregeneration after injury of the peripheral nervous system (PNS). Integrins are present at the growth cone of damaged PNS neurons and attach to ligands in the ECM to promote axon regeneration. It is unclear whether integrins can promote axon regeneration in the adult central nervous system (CNS).


It didn’t take long to get some answers about SARS-CoV-2.
From 2020 and in “Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26” , Vankadari & Wilce conducted an in silico modelling of spike glycoprotein homo-trimer binding to CD2611:

The docked complex model of COVID-19 spike glycoprotein and CD26 (Figure 2) shows a large interface between the proteins. This suggests a possible tight interaction between the S1 domain loops in the modelled structure and the CD26 surface.
Previous studies of CD26 binding have shown that residues K267, T288, A289, A291, L294, I295, R317, Y322 and D542 interact with Bat-CoV (MERS) spike protein [10].
Interestingly our docked model supports this despite the variability between these spike proteins’ S1 domains, with the same CD26 residues in close proximity to the active region of S1 domain in COVID-19. We also observed additional residues (Q286, I287, N338, V341, R336) of CD26 predicted to interact with the S1 domain of the spike protein via van der Waals or by hydrogen bonding. However, regarding the COVID-19 spike glycoprotein, we noticed many different and unique residues (R408, Q409, T445, V417, L461, D467, S469, L491, N492, D493, Y 494, T497, T150, Y504) predicted to interact with CD26. Some of these unique residues of S1 domain are also predicted interact with the ACE2 protein [6]. This underlines the novelty and uniqueness of COVID-19 and its interaction with human target proteins.
In contrast, this docking study from 2021 by Cameron et al concluded from their findings that DPP4 was not supported as being a significant receptor for SARS-CoV-2 unless the RBD of SARS-CoV-2 was in a different formation12 which is by no means unlikely, especially if expressed from synthetic mRNA. They also didn’t consider downstream effects on DPP4 after first binding to integrins.
“Does the SARS-CoV-2 Spike Protein Receptor Binding Domain Interact Effectively with the DPP4 (CD26) Receptor? A Molecular Docking Study”
Interactions that use neighbouring residues to those predicted by Li et al. include the residues K267, R336 and A291 of DPP4, with the residues Y499, E406 and Q493 of SARS-CoV-2. Li et al. predicted that these specific interactions occur between K267 and Q498, R336 and D405, and A289 and Q493 of DPP4 and SARS-CoV-2, respectively. In addition, the following interactions predicted by Li et al. were not reproduced in our simulations: Q344 and Y489, K392 and A475 and T478, Q286 and N501, and T288 and Y505 of DPP4 and SARS-CoV-2, respectively. Overall, the predicted binding interactions bear similarity to the predictions of Li et al. but do not reproduce their findings.
These findings indicate that the molecular simulations were unable to successfully dock the RBD of SARS-CoV-2 with DPP4 in spite of the conformational flexibility that HADDOCK allowed. This suggested that using a different initial conformation of the RBD of SARS-CoV-2 may be necessary, particularly since the conformation used was extracted from its crystal structure in complex with the ACE2 receptor.

Regardless of the models used, mutations may also select variants with higher docking affinities:
It is important to note that the interactions predicted in this docking study would likely be impacted in some of the new variants of SARS-CoV-2 that emerged in late 2020, such as those from the UK, South Africa, Brazil and India [18]. The UK variant (B.1.1.7 or Alpha) has the mutation N501Y in the RBD of SARS-CoV-2 [18]. This mutation is also present in the South African (B.1.351 or Beta) and Brazilian (P.1 or Gamma) variants, which also have the mutations K417N/T and E484K. The Indian (B.1.617.2 or Delta) variant has different mutations in the RBD of SARS-CoV-2: L452R and T478K. Whilst the N501Y and K417N/T mutations are known to favour the binding of SARS-CoV-2 to the ACE2 receptor [1,2,3], this docking study suggests that only N501Y, E484K and T478K are relevant to the best-predicted binding modes between SARS-CoV-2 and DPP4.
Apart from ex vivo clinical findings from the January ‘23 study by Brunetti et al, other evidence for binding to DPP4 comes from type 2 diabetes mellitus (T2DM) patients, as administration of DPP4 inhibitors (ie gliptins) alone was associated with improved clinical outcomes.
“Role of Dipeptidyl Peptidase-4 (DPP4) on COVID-19 Physiopathology13”:
It should be pointed out that the direct effect of DPP4 inhibitors on preventing coronavirus infection has not been demonstrated to date. However, increasing evidence illustrates that DPP4 inhibitors have a beneficial effect on the clinical outcome of patients by reducing COVID-19 complications, improving recovery, and reducing mortality.
In a multinational retrospective cohort study involving 56 large health care organizations, it was shown that the use of DPP-4 inhibitors was associated with a reduction in respiratory complications and a decrease in mortality, based on 2264 patients treated with DPP4 inhibitors only (i.e., alogliptin, linagliptin, saxagliptin, or sitgliptin) [141].
Likewise, a prospective randomized clinical trial with 263 COVID-19 patients showed that patients treated with sitagliptin for 2 days had better clinical outcomes and reduced lung infiltration than the control group [142].
Moreover, a prospective study with 89 COVID-19 but non-diabetic patients demonstrated that sitagliptin improved clinical outcomes, radiological scores, and inflammatory biomarkers, pointing to a potential usefulness of DPP4 inhibitors in managing non-diabetic COVID-19 patients [143]. A meta-analysis showed that the effect of gliptins was independent of age, sex, race, and location [144].
However, in most cases, DPP4 inhibitor users had other medications for T2DM like metformin, renin-angiotensin system inhibitors, thiazolidinediones, diuretics, or statin, making it difficult to attribute the beneficial effect solely to DPP4 inhibitors [145]. In spite of this, the clinical outcomes of COVID-19 patients using DPP4 inhibitors only was not significantly different from that using both DPP4 inhibitors and RAS inhibitors and was notably improved from COVID-19 T2DM patients without medication [145].
In 2021 Makowski et al considered integrins as alternative path for SARS-CoV-2 cell entry. Although they don’t mention Tat motifs by name, the whole paper refers to the arginine-glycine-aspartic acid (RGD) sequence which in the case of carboxyterminal of Tat helps mediate vascular cell migration and invasion by binding to the α5β1 and αvβ3 integrins, as above.
From “Biological and Clinical Consequences of Integrin Binding via a Rogue RGD Motif in the SARS CoV-2 Spike Protein”14 we learn more about the related pathologies that spike binding to integrins α5β1 or/and αvβ3 may induce, in addition to lymphopenia.
Key extracts:
This paper considers the hypothesis that viral binding to cell-surface integrins may contribute to the high infectivity and widespread extra-pulmonary impacts of the SARS-CoV-2 virus. This potential is suggested on the basis of the emergence of an RGD (arginine-glycine-aspartate) sequence in the receptor-binding domain of the spike protein.
Although RGD is a commonly used motif by viruses, they don’t all bind to CCR4, CCR5 and directly to CD4+ T cells. Two viruses now interact with all these, the other being HIV…
RGD is a motif commonly used by viruses to bind cell-surface integrins. Numerous signaling pathways are mediated by integrins and virion binding could lead to dysregulation of these pathways, with consequent tissue damage. Integrins on the surfaces of pneumocytes, endothelial cells and platelets may be vulnerable to CoV-2 virion binding. For instance, binding of intact virions to integrins on alveolar cells could enhance viral entry. Binding of virions to integrins on endothelial cells could activate angiogenic cell signaling pathways; dysregulate integrin-mediated signaling pathways controlling developmental processes; and precipitate endothelial activation to initiate blood clotting.
Ask yourself what conditions are more and more people, including previously healthy individuals being diagnosed with?
Such a procoagulant state, perhaps together with enhancement of platelet aggregation through virions binding to integrins on platelets, could amplify the production of microthrombi that pose the threat of pulmonary thrombosis and embolism, strokes and other thrombotic consequences.
Risk of immune escape and breakthrough infections due to selective pressure on viral strains through mass transfection campaigns:
Patient-specific differences in these factors may contribute to the high variability of clinical presentation. There is danger that the emergence of receptor-binding domain mutations that increase infectivity may also enhance access of the RGD motif for integrin binding, resulting in viral strains with ACE2 independent routes of cell entry and novel integrin-mediated biological and clinical impacts.
Although published in 2021 this type of mutation is an ongoing threat:
The highly infectious variant, B.1.1.7 (or VUI 202012/01), includes a receptor-binding domain amino acid replacement, N501Y, that could potentially provide the RGD motif with enhanced access to cell-surface integrins, with consequent clinical impacts.
This paper considers the hypothesis that integrins may provide an alternate path for SARS CoV-2 cell entry and that the frequently observed coagulopathy, angiogenesis and other clinical impacts observed during the severe second phase of COVID-19 infection are driven not only by the widely reported inflammatory response but, at least in part, by the direct interaction of virus particles with endothelial cells and platelets, among others. Coagulation and angiogenesis are cellular processes regulated, to some degree, by integrins. Evidence is considered here that suggests that an integrin-binding “RGD motif” near the distal tip of the SARS CoV-2 spike protein may interact with integrins on cell surfaces and lead to extensive dysregulation of these processes.
If integrins provide SARS CoV-2 with an ACE2-independent route to cell entry, the key interaction will be with the RGD motif in the receptor binding domain of the spike protein. The RGD (arg-gly-asp) tri-peptide motif is recognized by at least eight human integrins and binds to their extracellular domains with high affinity. Many human viruses use an RGD motif displayed on their virion surface to bind to the extracellular domain of integrins, thereby initiating cell entry and triggering dysregulated cellular signaling processes [18,19]. For instance, binding to integrin αIIbβ3, the principal platelet integrin, could dysregulate platelet aggregation. Initially promulgated through binding to the KQAGDV sequence in the γC region of fibrinogen, aggregation later involves binding of RGD sequences in the α chain of fibrin to integrin αIIbβ3. Similarly, virion binding to integrin αVβ3 could lead to dysregulation of angiogenesis since integrin αVβ3 is one of at least six RGD-binding integrins implicated in the regulation of vascular growth [20,21].
Experimental mRNA gene therapy agents were administered en masse from December 2020, and yet as at 2021 no-one had (at least publicly) investigated interactions with integrins:
In the absence of experimental data probing the affinity of the SARS CoV-2 spike protein to relevant human integrins, this paper explores the possibility that these interactions occur and are responsible for important clinical aspects of COVID-19.
1.1.2. Spike Protein Structure The spike protein of SARS CoV-2 virus contains an RGD motif near the distal tip of its receptor-binding domain [23] with structural features reminiscent of known integrin-binding proteins. Direct measurements of the potential binding of spike protein to relevant integrins have yet to be reported. In the absence of these critical data, this paper examines the structure of the RGD motif in the SARS CoV-2 spike protein and compares it with structures of RGD motifs on known integrin ligands and integrin-targeting virions.

Prior to binding, the FMDV RGD motif is part of a long, flexible loop [25,26] that is largely immobilized by binding to integrin.

Figure 2. Structure of known integrin-binding proteins: (A) Virion proteins known to bind integrins through an RGD motif (shown in space-fill) include (right) foot and mouth disease virus capsid protein (5neu—this RGD motif is highly flexible prior to integrin-binding, but structurally stabilized when bound to integrin—image is from a co-crystal of the capsid protein and integrin with integrin structure removed to make visible the RGD domain); (left) African horse sickness virus (1ahs—top domain of capsid protein VP7). (B) Other proteins known to bind integrin through an RGD motif: thrombospondin (1ux6); prothrombin (3u69); rhodostomin (4rqg) and triflavin (1j2l) are disintegrins, small toxins from snake venom with high affinity to integrins; and fibronectin (1fnf—domains 6–10)—an extracellular matrix protein with an integrin-binding RGD motif in its 10th domain. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7909284/ 


The structure of two disintegrins, rhodostomin (4rqg) and triflavin (1j2l), small protein components of viper venom that bind tightly to integrin αIIbβ3 on platelets, can be seen in Figure 2B to have RGD motifs that extend from the body of the protein to facilitate interaction with integrins. These toxic proteins act as potent anti-coagulant drugs by blocking fibrinogen-binding to integrin αIIbβ3. Naturally occurring integrin ligands are also depicted in Figure 2B, including thrombospondin (1ux6), prothrombin (3u69) and fibronectin (1fnf), all exhibiting RGD motifs that extend from the body of the protein. Generally, the RGD motif of naturally occurring integrin ligands is either highly exposed on the protein surface, highly flexible or both. This would suggest that efficient binding of a spike protein to an integrin could be dependent on a microenvironment conducive to the disordering of the extreme distal tip of the spike protein. Small variation in microenvironmental conditions could lead to large fluctuations in the availability of the RGD motif for integrin-binding and lead to consequent variability in clinical manifestations.
Of note here that clotting disorders were previously discussed in the context of HIV and Tat, to take us full circle:
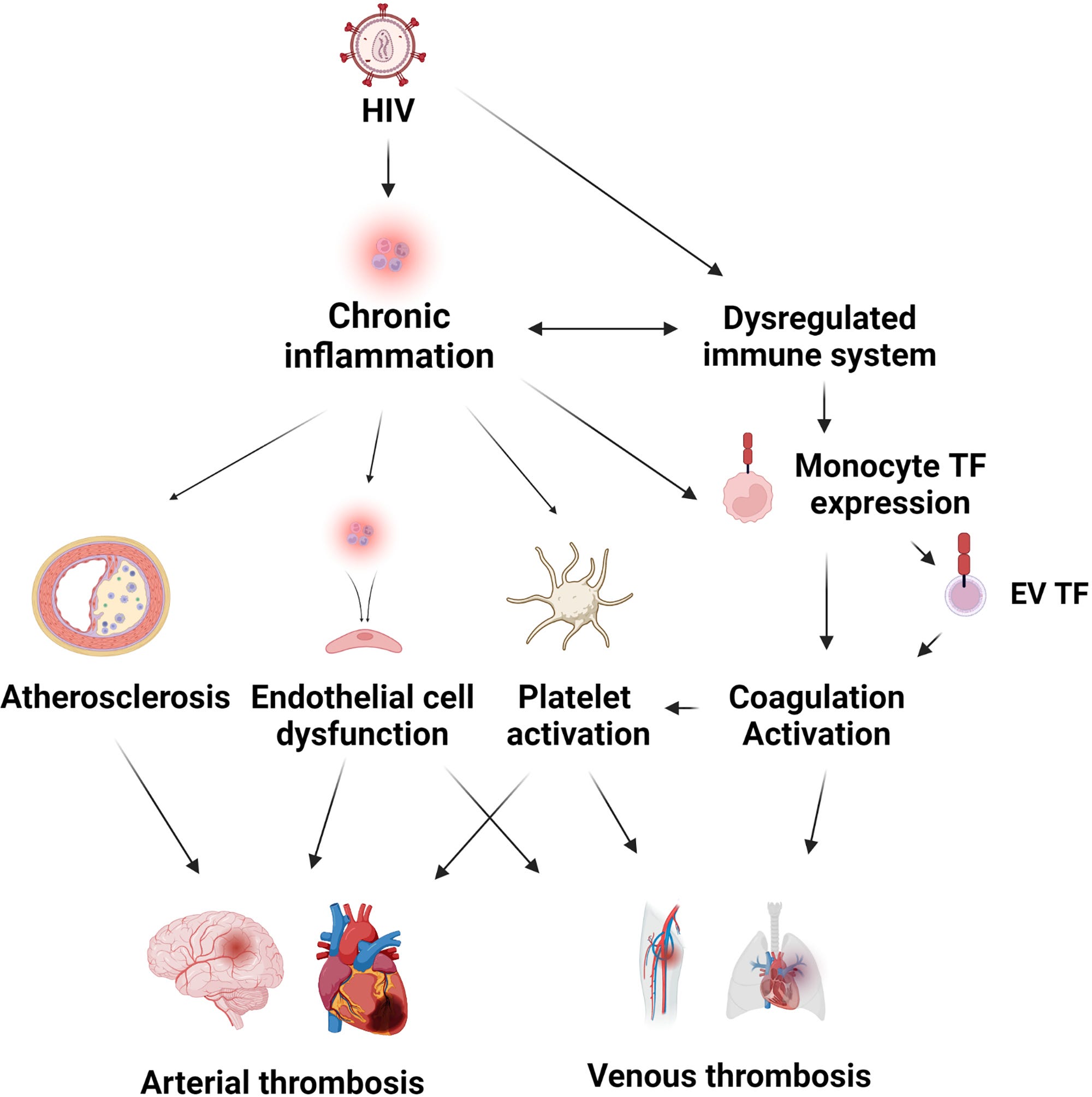
“Cardiovascular Disease and Thrombosis in HIV Infection” (2022)
Cardiovascular disease and thrombosis have become an increasing problem for people living with HIV, and the burden will continue to increase as more people become infected.
People living with HIV are at an increased risk for arterial and venous thrombosis.


Fibronectin has numerous functions that ensure the normal functioning of vertebrate organisms. It is involved in cell adhesion, growth, migration, and differentiation. Cellular fibronectin is assembled into the extracellular matrix, an insoluble network that separates and supports the organs and tissues of an organism.
Fibronectin plays a crucial role in wound healing. Along with fibrin, plasma fibronectin is deposited at the site of injury, forming a blood clot that stops bleeding and protects the underlying tissue. As repair of the injured tissue continues, fibroblasts and macrophages begin to remodel the area, degrading the proteins that form the provisional blood clot matrix and replacing them with a matrix that more resembles the normal, surrounding tissue. Fibroblasts secrete proteases, including matrix metalloproteinases, that digest the plasma fibronectin, and then the fibroblasts secrete cellular fibronectin and assemble it into an insoluble matrix. Fragmentation of fibronectin by proteases has been suggested to promote wound contraction, a critical step in wound healing.
Role in cancer
Several morphological changes has been observed in tumors and tumor-derived cell lines that have been attributed to decreased fibronectin expression, increased fibronectin degradation, and/or decreased expression of fibronectin-binding receptors, such as α5β1 integrins.
Fibronectin has been implicated in carcinoma development. In lung carcinoma, fibronectin expression is increased especially in non-small cell lung carcinoma. The adhesion of lung carcinoma cells to fibronectin enhances tumorigenicity and confers resistance to apoptosis-inducing chemotherapeutic agents. Fibronectin has been shown to stimulate the gonadal steroids that interact with vertebrate androgen receptors, which are capable of controlling the expression of cyclin D and related genes involved in cell cycle control. These observations suggest that fibronectin may promote lung tumor growth/survival and resistance to therapy, and it could represent a novel target for the development of new anticancer drugs.
Fibronectin 1 acts as a potential biomarker for radioresistance and for pan-cancer prognosis.
FN1-FGFR1 fusion is frequent in phosphaturic mesenchymal tumours.
Virus particles that display “RGD” on their surface invariably utilize integrins as sites of attachment and cell entry and, in many cases, also trigger signal pathways that control aspects of cellular development, growth and motility.
There are three ways in which the binding of SARS CoV-2 to an integrin could impact tissue integrity. The simplest (Figure 6 top) is for the virion to bind and act as an antagonist, a competitive inhibitor that blocks the interaction of the integrin with a ligand, disrupting signaling and downstream cellular responses due to that ligand.
The second way (Figure 6, middle) is for the virion to act as an agonist, triggering signaling pathways and leading to dysregulated and potentially damaging cellular responses.
The third way (Figure 6, bottom) is to utilize the integrin as a portal for cell entry, leading to viral replication and cellular disruption. In the first two cases, the virion may remain attached to the cell surface as a multivalent adhesive element that could contribute to uncontrolled cell–cell adhesion. If bound to platelets, this could contribute to platelet aggregation and the coagulopathies frequently observed in COVID-19.

6. Integrins, SARS CoV-2 and Angiogenesis
Although the induction of angiogenesis is most often associated with the action of vascular endothelial growth factor (VEGF), at least six integrins, αVβ3, αVβ5, α5β1, α2β1, αVβ1 and α1β1, have been implicated as contributors [20,21]. If the spike protein of SARS CoV-2 exhibits affinity for integrins, it may be capable of triggering dysregulated angiogenesis.
The disruption of pulmonary tissue resulting from uncontrolled angiogenesis contributes to the extensive lung damage observed in the most severe cases. For SARS CoV-2 virions to induce processes of this kind, the RGD motif on the spike protein would need to act as an agonist, activating, among others, integrins αVβ3 and/or αVβ5, long considered to be positive regulators of the angiogenic switch [61,62]. This is not unprecedented—activation of signaling pathways by the interaction of virion-displayed RGD motifs with integrins is relatively common in human viruses.
This is how Kaposi’s sarcoma-associated virus can induce tumorigenesis, but they don’t actually mention this or carcinogenesis:
For instance, Kaposi’s sarcoma-associated virus (KSHV) utilizes an RGD motif on surface glycoprotein gB to bind host cell integrins, gain cell entry [44] and act as an agonist to activate numerous signaling pathways [63]. KSHV triggers the association of integrins with Rho GTPases, tyrosine kinase receptors and Toll-like receptors that results in cytoskeletal remodeling, differential cell type targeting and innate responses [64]. These activities are mediated through at least three integrins [45]. It represents a prime example of how the interaction of RGD motifs with integrins can lead to virally induced activation of integrin-mediated pathways.

And there are case reports for this. The authors speculate that reactivation of latent viruses are a key contributory factor.
From 2021 by Magri et al, “New-onset cutaneous kaposi’s sarcoma following SARS-CoV-2 infection”15.
Key extracts:
Background. COVID-19 is associated with several cutaneous manifestations, including chilbain-like lesions, urticaria, erythema multiforme, and maculopapular lesions. Dermatoses may be directly linked to the viral infection or also represent a consequence of systemic therapies administrated for COVID-19. A potential role of SARS-CoV-2 in triggering the reactivation of other viruses, such as HHV-6, HHV-7 and Epstein-Barr virus has been hypothesized.
Objective. To better understand and hypothesize possible pathogenetic correlations of COVID-19 with other dermatological conditions.
Methods. We report the case of an 83-year-old woman hospitalized in a nursing home for several years. On November 2020, the patient had been diagnosed with SARS-CoV-2 infection, with repeated positive swabs until January 2021. After a month, new-onset asymptomatic cutaneous purplish macular lesions and violaceous patches occurred bilaterally on the feet.
Results. An incisional cutaneous biopsy and the histological examination of the plantar lesion revealed the diagnosis of Kaposi Sarcoma.
Conclusion. We report a unique case of new-onset bilateral Kaposi's sarcoma following COVID-19, speculating on a possible role of SARS-CoV-2 in the reactivation of human herpes virus-8 (HHV-8) infection.
Interestingly, Chen et al. have recently reported that SARS-CoV-2-encoded proteins and anti-COVID-19 drugs are able to induce lytic reactivation of KSHV, suggesting that in KSHV+individuals, especially in endemic areas, the exposure to coronavirus may increase the risk to develop virus-related tumors.12
Misdiagnosis may mean the true incidence is being under reported:
Besides the pathogenic implications, another interesting aspect of our case is its possible differential diagnoses. Generally, the purplish-violaceous lesions of KS may be confused with other conditions, such as ecchymosis, bacillary angiomatosis, hemosiderotic hemangioma, pyogenic granuloma, and pseudomyogenic hemangioendothelioma.13, 14 Furthermore, KS may mimic chilblains,15 and chilblain-like lesions have been largely described as a COVID-19 skin manifestation. In our case, the initial onset of roundish violaceous spots on the feet and especially on the posterior region of the fingers could be easily confused with this frequently reported sign of COVID-19. Thus, physicians working in COVID-19 hospital should be aware of this possible, even if uncommon, differential diagnosis of chilblain-like lesions.
HIV also mediates Kaposi’s sarcoma via reactivation of HHV-816.
It is somewhat ironic that research into HIV induced pathology and therapeutics is much more comprehensive than comparative studies into SARS-CoV-2 and related transfection agents, and yet this huge body of relevant research has been almost completely ignored.
H*V is almost verboten to discuss in relation to SARS-CoV-2 and transfection agents. This is partly why I’m writing these reviews. Ignoring the presence of the “borrowed” motifs doesn’t make the pathology go away or help you to diagnose and treat the patients effectively:
“Integrin αVβ3 as a Target for Blocking HIV-1 Tat-Induced Endothelial Cell Activation In Vitro and Angiogenesis In Vivo” (2005)
Objective— The transactivating factor (Tat) of HIV-1 binds to αvβ3 integrin present on endothelial cells contributing to neovascularization. Here, we investigated the biological consequences of Tat/αvβ3 interaction and the antagonist effect of an Arg-Gly-Asp (RGD)-based peptidomimetic.
Conclusion— These data provide new insights on the mechanism of endothelial cell activation by Tat and point to RGD peptidomimetics as prototypes for the development of novel Tat antagonists.
The transactivating factor (Tat) of HIV-1 binds to endothelial αvβ3 integrin triggering focal adhesion kinase and NF-κB activation that lead to endothelial cell proliferation, membrane ruffling, and motility in vitro and neovascularization in vivo. The RGD-peptidomimetic SCH221153 inhibits these biological activities with high efficiency and specificity.
Tat protein, the main transactivating factor of HIV-1,1 is released by HIV-1–infected cells2 targeting different types of uninfected cells and causing a variety of biological effects related to distinct AIDS-associated pathologies. In AIDS patients, Tat contributes to tumorigenesis,2 to the pathogenesis of dementia,3 to the immune system suppression (by interfering with the function of different cells of immunity),2 and to heart disease and atherosclerosis.4
The involvement of endothelial cells (ECs) in Tat-dependent pathologies is manifold.5 By inducing neovascularization, Tat contributes to tumor progression and Kaposi sarcoma (KS) development.2 Also, Tat increases endothelial permeability and alters the expression of endothelial leukocyte receptors, leading to the extravasation of HIV-1+ monocytes and HIV-1 dissemination. These effects, in turn, contribute to the pathogenesis of lymphomas, AIDS-dementia,5 cardiovascular diseases, and atherosclerosis.6 A decrease in endothelium-dependent vasorelaxation and endothelial nitric oxide synthase expression may also play a role in endothelial dysfunctions mediated by Tat.7
On these bases, Tat is considered a main target for the development of anti-AIDS therapies, and different anti-Tat strategies have been described, including gene therapies, vaccines, and Tat antagonists.8
Tat accumulates in the extracellular matrix as an immobilized protein,9 and substrate-immobilized Tat interacts with αvβ3 integrin of ECs promoting their adhesion and triggering a complex signal transduction pathway that leads to activation of Tat-adherent ECs.10 Accordingly, αvβ3 interaction is required for the chemotactic and mitogenic activity of Tat in vitro and for its angiogenic activity in vivo.11 The Arg-Gly-Asp (RGD) tripeptide present in position 78 to 80 in Tat protein is required for αvβ3 interaction and EC adhesion.11 Accordingly, RGD-containing peptides inhibit cell adhesion to immobilized Tat.12
https://www.ahajournals.org/doi/full/10.1161/01.ATV.0000186182.14908.7b
Integrins and cancer
On conducting a search of the journals, integrin α5β1 is strongly correlated with cancer. We have already seen that binding of both HIV and spike protein motifs to α5β1 and αvβ3 activates them, leads to pathologies including thrombocytopenia and endothelial damage due to neovascularisation. Carcinogenesis is a related consequence of this.
For example, urological tumours are associated with activated α5β1 and inhibiting it presents a potential treatment, especially for otherwise resistant tumours.
From Zhou et al (2023), “Targeting integrin α5β1 in urological tumors: opportunities and challenges”17.
Key extracts:
Urological tumors, such as prostate cancer, renal cell carcinoma, and bladder cancer, have shown a significant rise in prevalence in recent years and account for a significant proportion of malignant tumors. It has been established that metastasis to distant organs caused by urological tumors is the main cause of death, although the mechanisms underlying metastasis have not been fully elucidated.
The fibronectin receptor integrin α5β1 reportedly plays an important role in distant metastasis and is closely related to tumor development. It is widely thought to be an important cancer mediator by interacting with different ligands, mediating tumor adhesion, invasion, and migration, and leading to immune escape.
The α-subunits are mainly associated with receptor recognition and contribute to binding integrin receptors with cation-dependent fits. The β-subunits are associated with cell-to-mesenchyme and cell-to-cell signaling and are involved in cytoskeletal protein interactions and intracellular signaling (8, 10, 11).
The integrin α5 subunit usually binds to the β1 subunit to form a heterodimeric integral membrane protein, the only known α5 integrin (18). After binding to the integrin α5β1 cytoplasmic tail and associated ligands, it binds to the cytoskeleton and drives cytoskeletal reorganization through an outside-in signaling pathway.
Integrin α5β1-regulated intracellular signaling activates the extracellular compartment and assists in assembling the extracellular matrix, i.e., an inside-out signaling pathway (19). This bidirectional signaling pathway involves biological behaviors such as cell adhesion, migration, and survival (20).
In addition, integrin α5β1 can act as a pro-angiogenic factor that is involved in tumor angiogenesis by interacting with vascular endothelial growth factor receptor and angiopoietin and has received a great deal of attention for its importance in tumorigenesis, metastasis, and drug resistance (21).
Prostate cancer:
It has been established that, in prostate cancer, there is a correlation between altered integrin expression and abnormal extracellular matrix secretion, progression, and invasion (42, 43). Several studies have reported the dysregulation of both integrin α and β subunits during prostate cancer progression (44, 45). Fibronectin polymerization is an important regulator of extracellular matrix stability (25, 46). A study revealed that the blockade of integrin α5β1 with the proline–histidine–serine–arginine–asparagine (PHSCN) peptide significantly prevented cell metastasis in preclinical prostate cancer models and in phase I clinical trials conducted in parallel (47).
RGD again, and the effects of competitive inhibition:
Moreover, the blockade of integrins using the fibronectin-related peptide GRGDSP (Gly-Arg-Gly-Asp-Ser-Pro) completely inhibited the growth and cell morphological alterations of prostate cancer PC-3 cells, confirming that integrins interact with the FNIII10 structural domain and play a key role in these processes (48, 49).
The inhibition of the α5 subunit in vivo has been shown in other studies to significantly inhibit tumor growth (52, 53). In conclusion, integrin α5 plays an important role in prostate cancer progression, and it can be inferred that integrin α5-related inhibitors may contribute to blocking tumor progression.
Bladder cancer:
Integrin α5β1 has also been correlated with the development and progression of bladder cancer.
Laidler et al. (56) found that the expression of integrin subunits α5 and β1 was significantly higher in malignant bladder cancer cells Hu456 and T24 than in non-malignant uroepithelial cell HCV29, suggesting that changes in integrin α5β1 expression may also contribute to bladder metastasis cell carcinoma progression, invasion, and metastasis.
Kidney cancer:
Integrin α5 is expressed at significantly higher levels in renal clear cell carcinoma than in normal tissue and plays an important role in renal cancer progression (62).
Haber et al. (64) found that increasing integrin α5 levels and downstream signaling through AKT could help tumor cells adhere to extracellular matrix compounds and promote bone metastasis in renal cell carcinoma.
Mechanisms. Note the RGD binding site:
Integrin α5β1 does not directly regulate tumor proliferation, metastasis, and drug resistance but by a combination of extracellular ligands and intracellular signaling pathways. Some mechanistic studies on integrin α5β1-mediated urological tumors in related aspects are briefly described below (Figure 1).

Figure 1. Schematic diagram of the signaling pathways involved in integrin α5β1 that mediate tumor cell proliferation, migration, angiogenesis, and drug resistance. Integrin α5β1 contributes to cancer progression by activating the PI3K/AKT, MAPK, and ERK signaling pathways. LOXL2 protein regulates the stability of integrin α5β1, which is also the downstream target of many miRNAs. Integrin α5β1 is also involved in mTOR inhibitor resistance through the FAK/Src axis. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10358839/

Drug resistance. If your chemotherapy is no longer as effective after exposure to spike protein, this could be a contributory factor:
During clinical practice, drug resistance is commonly observed in the treatment of tumors, and studies have shown that integrin α5β1 is involved in this phenomenon. Juengel et al. found that integrin α5 was involved in the development of renal clear cell carcinoma resistant to mTOR inhibitors, characterized by quantitative changes in integrin α5 expression during drug resistance and coupled with altered molecular function of integrins, forcing renal clear cell carcinoma to shift from adhesion to migration (88).
Similarly, in studies of resistance to mTOR inhibitors in prostate cancer, β1 integrins significantly triggered the migration of tumor cells, mediating the activation of the AKT signaling pathway and triggering cancer cell metastasis during the upregulation of β1 integrin expression (89).
In conclusion:
Dysregulation of integrin α5β1 is strongly related to the development of urological tumors and may serve as an important indicator for evaluating invasion and migration. Additionally, the upregulation of integrin α5β1 can promote the development of drug resistance in cancer cells, which prompted the exploration of novel strategies for overcoming drug resistance in chemotherapy. The overexpression of integrin α5β1 has been documented in various tumors and reportedly contributes to tumor progression, making it a potential target for tumor imaging and an independent indicator of poor prognosis.
Integrins are cell surface adhesion (sticking) proteins that allow cells to interact with the environment. In cancer these proteins can "misbehave" and this can contribute to the development of cancer, and to cancer spreading (metastasis).
Adapted from: https://www.youtube.com/watch?v=p-zlBgs1p3A
As an integrin with α and β subunits, as with α5β1, αvβ3 has also been shown to promote aggressive phenotypes in many types of cancers, including prostate18, breast19, bone20, brain (glioblastoma)21 and lung22.
Antiplatelet autoimmunity
In 2018 Zeng et al published a study where they analysed the venous blood and bone marrow biopsies of 103 patients who had been diagnosed with chronic immune thrombocytopenia (ITP):
“What is ITP?”
Immune thrombocytopenia (ITP) is a type of platelet disorder. In ITP, your blood does not clot as it should, because you have a low platelet count. Platelets are tiny blood cells that are made in the bone marrow. When you are injured, platelets stick together to form a plug that seals your wound. This plug is called a blood clot. When you have a low platelet count, you may have trouble stopping bleeding.
ITP can be acute (short-term) or chronic (long-term). Acute ITP often lasts less than 6 months. It mainly occurs in children — both boys and girls — and is the most common type of ITP. Chronic ITP lasts 6 months or longer and mostly affects adults. However, some teens and children do get this type of ITP. Chronic ITP affects women two to three times more often than it affects men.
What are the symptoms?
ITP may not cause any symptoms. However, ITP can cause bleeding that is hard to stop. This bleeding can be inside your body, underneath your skin, or from your skin.
Signs of bleeding may include:
Petechiae, which are small, flat red spots under the skin caused by blood leaking from blood vessels
Purpura, which is bleeding in your skin that can cause red, purple, or brownish- yellow spots
Clotted or partially clotted blood under your skin (called a hematoma) that looks or feels like a lump
Nosebleeds or bleeding from your gums
Blood in your urine or stool
Heavy menstrual bleeding
Extreme tiredness
The biopsy samples were then analysed using a range of tests:
Modified antigen capture ELISA (MACE) for detection of anti-αvβ3 autoantibody.
Flow cytometry analysis.
Trypan blue staining to calculate cell death rate.
Cell viability assay.
Adhesion analysis.
Migration analysis.
Western blotting for proteins.
Immunofluorescence staining with antibodies against p-FAK and p-SRC.
Proplatelet formation assay.
A separate ITP model mouse experiment was conducted using various antibodies.
A hematological parameter test to quantify the platelet count.
Their analysis led to the following findings, ie the pathophys behind the clotting disorders:
The total positive rates of anti-αIIbβ3 autoantibody and anti-αvβ3 autoantibody detected were 35.92% (n = 37) and 30.10% (n = 31), respectively. Among 31 anti-αvβ3-positive patients, 29 patients were also anti-αIIbβ3 positive.
A megakaryocyte (mega- + karyo- + -cyte, "large-nucleus cell") is a large bone marrow cell with a lobated nucleus that produces blood platelets (thrombocytes), which are necessary for normal clotting. In humans, megakaryocytes usually account for 1 out of 10,000 bone marrow cells, but can increase in number nearly 10-fold during the course of certain diseases. Owing to variations in combining forms and spelling, synonyms include megalokaryocyte and megacaryocyte.

...we examined the pathological changes in the BM biopsies and observed no significant decrease in the total number of MKs in chronic ITP patients. Interestingly, compared with the anti-αvβ3 autoantibody-positive group, more megakaryocytes were predominantly located in the vicinity of sinusoidal BMECs in the anti-αvβ3 autoantibody-negative group.
Anti-αvβ3 antibody does not directly influence the survival and proliferation of MKs but impedes proplatelet formation.
Compared with the control group, proplatelet formations significantly decreased in LM609-treated MKs (Fig. 2G, H). The reduced proplatelet formation in MKs after anti-αvβ3 antibody treatment was further confirmed by immunofluorescent anti-tubulin staining.
Anti-αvβ3 antibody inhibits MK migration via suppressing AKT activation. The migration of MKs from the osteoblast niche to the vascular niche is a key event in thrombocytopoiesis in vivo, and SDF-1 plays a crucial role in regulating MK migration.
Anti-αvβ3 antibody inhibits MK adhesion via blocking the phosphorylation of FAK and SRC. Adhesion to bone marrow stroma is crucial for MK migration and is also a key event in thrombocytopoiesis.
Anti-αvβ3 antibody impedes MK migration and adhesion to vascular endothelial cells…These data demonstrate that anti-αvβ3 antibody can impede MKs’ migration and adhesion to ECs, which may result in the delayed release of platelets into the vascular sinus.
Anti-αv antibody injection delays platelet recovery in the anti-αIIb antibody-induced ITP mouse model.
GP-specific IgGs, such as αIIbβ3 autoantibodies, have been widely recognized and thought to be the primary causes of ITP 1, 6, 8. However, the pathological significance of the anti-αvβ3 autoantibody in ITP patients has not been thoroughly elucidated to date. In this study, we demonstrated that the anti-αvβ3 autoantibody aggravates thrombocytopenia by impeding MK migration and adhesion to vascular endothelial cells, probably through suppression of cellular signals mediated by AKT, FAK and SRC.
Taken together, these data partially demonstrate that integrin αvβ3 may be involved in the regulation of thrombocytopoiesis, and the anti-αvβ3 autoantibody is likely to be synergistic with the anti-αIIbβ3 autoantibody and contributes to the pathogenesis of refractory ITP by inhibiting the migration and adhesion of MKs to the vascular niche.
Other studies into antiplatelet autoimmunity
CITP:

{kind=link}
#/media/File:6CYT_HIV_Tat_TAR_complex.png){kind=link}
{kind=link}
{kind=link}
“Viral-Associated Immune Thrombocytopenic Purpura” (2008)
Chronic immune thrombocytopenic purpura (CITP) is a diagnosis of exclusion that occurs either de novo or secondary to other underlying disorders. Chronic infection with human immunodeficiency virus (HIV) and hepatitis C virus (HCV) are now well-characterized causes of CITP. Between 6% and 15% of patients infected with HIV may develop thrombocytopenia. Patients with CITP with risk factors for HIV infection should be screened for the virus.
A study into SARS-CoV-2 vaccines and antiplatelet autoimmunity from 2022 by Petito et al:
“Anti-severe acute respiratory syndrome coronavirus-2 adenoviral-vector vaccines trigger subclinical antiplatelet autoimmunity and increase of soluble platelet activation markers23”
Key extracts:
To slow down the coronavirus disease 2019 (COVID-19) pandemic an unequalled vaccination campaign was initiated. Despite proven efficacy and safety, a rare but potentially fatal complication of adenoviral-vector vaccines, called vaccine-induced immune thrombotic thrombocytopenia (VITT), has emerged the pathogenesis of which seems to be related to the development of platelet-activating anti-platelet factor 4 (PF4) antibodies.
Again, no studies had been conducted into antiplatelet responses and the risk of clotting until long after the rollout of experimental gene therapy agents:
While a few studies have evaluated the incidence of anti-PF4 positivity in anti-severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) vaccine recipients, to date no studies have assessed whether an antiplatelet immunological response develops and if this associates with platelet and blood clotting activation. We carried out a prospective study in healthy subjects who received the first dose of ChAdOx1 or Ad26.COV2.S or BNT162b2 vaccines to evaluate platelet-specific and non-specific immune response and in vivo platelet activation and blood clotting activation.
Individuals receiving ChAdOx1 and, less so, Ad26.COV2.S developed with high frequency auto- or alloantiplatelet antibodies, increased circulating platelet-derived microvesicles and soluble P-selectin associated with mild blood clotting activation.
And yet the products weren’t instantly recalled, and still haven’t been, over a year after this was published:
Our study shows that an immunological reaction involving platelets is not uncommon in individuals receiving anti-SARS-CoV-2 vaccination, especially after ChAdOx1 and Ad26.COV2.S, and that it associates with in vivo platelet and blood clotting activation.
Only a few investigations to date have assessed the impact of adenoviral-vector and mRNA anti-SARS-CoV-2 vaccines on platelets and the blood clotting system in apparently healthy individuals.
However, to date no studies have assessed whether an immunological response involving platelets develops in healthy subjects after adenoviral-vector or mRNA-based vaccine administration and if this associates with platelet activation.
We carried out a prospective study in healthy subjects who received the first dose of ChAdOx1, Ad26.COV2.S or BNT162b2 vaccines to evaluate platelet-specific and non-specific immune response, in vivo platelet activation and blood clotting activation.
A total of 72 subjects undergoing first dose ChAdOx1, Ad26.COV2.S or BNT162b2 administration (ChAdOx1, n = 32; Ad26.COV2.S, n = 20; BNT162b2, n = 20) were enrolled in an observational prospective study approved by the local Ethics Committee (Bioethics Committee, University of Perugia, approval n. 83 886).
In this study TAT = “Thrombin-antithrombin complex”
Average fibrinogen levels were within reference intervals (200–400 mg/dl) both before (T0) and after vaccine administration (T1) in all groups. However, in subjects receiving ChAdOx1 and BNT162b2 a significant decrease of plasma fibrinogen was observed at T1 compared to T0 (Table 2), with 65.6% and 30% of subjects respectively showing levels below the lower limit.
There were no significant differences among the three groups for TAT levels either at T0 or at T1. However, in subjects receiving Ad26.COV2.S and BNT162b2 a significant increase of plasma TAT was observed at T1 compared to T0 (Table 2), while no significant increases at T1 were observed in any of the three groups for F1 + 2 levels (Table 2).
Anti-PF4/heparin antibodies:
In the Ad26.COV2.S group positivity for IgG/IgA/IgM anti-PF4/heparin antibodies was detected at T1 in one subject (5%) (weakly positive, OD <0.42; Table S2A), as well as IgG anti-PF4/heparin antibody positivity (5%) (weakly positive, OD <0.5; Table S2B).
In the BNT162b2 group no subject was positive for IgG/IgA/IgM anti-PF4/heparin, while IgG anti-PF4/heparin antibodies were detected in four subjects at T1 (21%) (only one highly positive, OD = 0.888), two of whom were positive also before vaccine administration (one highly positive, OD = 1.147, the same patient highly positive also at T1; Table S2B).
Platelet auto- and alloantibodies. These are highly significant numbers IMOH:
Antiplatelet autoantibody positivity at T0 was found in a small and similar number of subjects in the three groups (<10%), while new positivity at T1 was found in 34% of subjects in the ChAdOx1 group (none of them had thyroid disease as a comorbidity), in 25% of those in the Ad26.COV2.S group and in 5% of those in the BNT162b2 group (ChAdOx1 vs. BNT162b2, p < 0.05; Table 3).

Antiplatelet alloantibody positivity at T0 was found in a similar number of subjects in the three groups, while new positivity at T1 developed in six ChAdOx1-vaccinated subjects (18.7%), in six Ad26.COV2.S-vaccinated subjects (30%), and in two BNT162b2-vaccinated subjects (10%; Table 3).
Microvesicles, also known as microparticles, are small extracellular vesicles released by cells in response to activation or apoptosis. Among the different microvesicles present in the blood of healthy individuals, platelet-derived microvesicles (PMVs) are the most abundant.
…Similarly to platelets, PMVs are also involved in thrombosis through support of the coagulation cascade. The levels of circulatory PMVs are altered during several disease manifestations such as coagulation disorders, rheumatoid arthritis, systemic lupus erythematosus, cancers, cardiovascular diseases, and infections, pointing to their potential contribution to disease and their development as a biomarker.
Incubation of whole blood from healthy subjects with ChAdOx1, but not with BNT162b2, induced a significant increase in the release of platelet-derived microvesicles (Figure 2).

Discussion:
Our study shows that the development of an immunological reaction involving platelets is not uncommon in subjects receiving anti-SARS-CoV-2 vaccination, but it highlights also that this is significantly more frequent in those vaccinated with ChAdOx1 and, although less commonly, with Ad26.COV2.S, and most importantly, that this is associated with in vivo platelet activation.
Similarly, hints to mild in vivo blood clotting activation were observed in a significant fraction of subjects receiving adenoviral-vector vaccines but not in BNT162b2-vaccinated subjects.
A sin by omission, they failed to discuss the “65.6% and 30% of subjects respectively showing levels below the lower limit” of plasma fibrinogen after administration of either BNT162bt2 or ChAdOx1. This means that your haemostatic coagulation system may fail and your blood will not clot correctly if exposed to trauma24.
As for the new incidence of anti-PF4/heparin antibodies in a significant percentage of individuals it would certainly have clinical relevance if you are an edge case or in a vulnerable group:
Low titre anti-PF4/heparin antibody positivity was previously reported in 1.05%–7.0% of healthy individuals who did not receive heparin,23, 39 further suggesting that low titre non-platelet-activating anti-PF4/heparin antibodies found after vaccine administration do not have any clinical relevance.
Imagine this in your brain or vital organs. A marker for disease, this was after only one dose and autoimmune antibody formation may be cumulative upon repeat exposure to an antigen:
On the other hand, adenoviral vector vaccine-elicited antiplatelet auto- and alloantibody positivity in our study was associated with in vivo platelet activation, documented by raised circulating platelet-derived microvesicles and sP-sel, and mild in vivo blood clotting activation. The occurrence of autoimmune phenomena, and among these of immune thrombocytopenic purpura, as adverse events of different vaccines is a well-know phenomenon.40
Transient thrombocytopenia with glycoprotein-specific platelet autoantibodies after adenoviral-vector anti-SARS-CoV-2 vaccination has been reported, showing that adenoviral-vector vaccination may raise a transient production of platelet-specific autoantibodies or of anti-viral antibodies cross-reacting with platelet glycoproteins in certain individuals.42
Anti-idiotypic antibodies may, through molecular mimicry, show sequence homology to the original antigen. AIA’s with homology to spike protein can be induced by reacting to anti-ACE2 antibodies, providing in principle an almost unlimited number of spike protein like AIA’s that are capable of binding ACE225, or in this case platelet related factors.
“Anti-idiotypic antibodies: biological function and structural studies” (1995)
Under a variety of circumstances antibodies can be elicited against the variable region of other antibody molecules (anti-idotypic antibodies, anti-ids). Some of the antibodies are directed against the binding sites of the eliciting antibodies. Of particular interest are the antibodies that recognize epitopes of the original antibody that are in contact with antigen.
…For an epitope that is sequence-specific (anti-FIPV system), molecular mimicry appears to be present as evidenced by the sequence homology between the CDR loops of the anti-id and the epitope of the original antigen. In the case of a small hormone antigen (angiotensin II), an internal image of the eliciting antigen appears to be represented in a single CDR loop of the antiiodiotypic antibody.
https://faseb.onlinelibrary.wiley.com/doi/10.1096/fasebj.9.1.7821758
Moreover, vaccine-elicited anti-spike protein specific antibodies (Ab1) have immunogenic amino acid sequences, called idiotopes, that can elicit antibody responses called anti-idiotype antibodies (Ab2). Ab2 antibodies can bind to the protective neutralising Ab1 antibody, thus impairing Ab1 efficacy, but they can also mirror the spike protein itself binding its target, the angiotensin-converting enzyme 2 (ACE2) receptor, possibly resulting in pathological effects.43 The SARS-CoV-2 spike protein has been suggested to bind either ACE-244 or glycoproteins such as αVβ3 and α5β145 on platelets thus activating them, and vaccine-elicited anti-idiotype antibodies might turn on platelets through this mechanism.
Therapeutics
This is somewhat challenging as receptor binding and autoimmune disorders are difficult to treat without using allopathic meds and risking side effects, eg DPP4 inhibitors (gliptins), as taken by diabetics26:
Gastrointestinal problems – including nausea, diarrhoea and stomach pain.
Flu-like symptoms – headache, runny nose, sore throat.
Skin reactions – painful skin followed by a red or purple rash.
Viral & spike protein reduction, proinflammatory cytokine reduction and immune support are easier to achieve though, i.e. damage limitation and for preventative purposes:
Even baicalin doesn’t appear to be able to directly prevent thrombocytopenia, and it blocks many many pathological signalling pathways:
However a type that can help to treat resultant thrombocytopenia are ellagic acids.
Efficacy is due to the promotion of megakaryocyte differentiation and enhanced platelet count as a result.
We have to be careful here though as the PI3K-Akt signalling pathway is being upregulated, but other significant cancer pathway blocking effects of ellagic acid will counter the risk of off-target effects2728.
“Two Ellagic Acids Isolated from Roots of Sanguisorba officinalis L. Promote Hematopoietic Progenitor Cell Proliferation and Megakaryocyte Differentiation” (2014)
The ellagic acids significantly stimulated the proliferation of Baf3/Mpl cells. Morphology analysis and megakaryocyte specific-marker CD41 staining confirmed that the ellagic acids induced megakaryocyte differentiation in HEL cells.
…This is the first time that 3,3',4-tri-O-methylellagic acid or 3,3',4-tri-O-methylellagic acid-4'-O-β-d-xyloside are reported to induce megakaryopoiesis, suggesting a class of small molecules which differ from others non-peptidyl, and appears to have potential for clinical development as a therapeutic agent for patients with blood platelet disorders.
“DMAG, a novel countermeasure for the treatment of thrombocytopenia” (2021)
In this study, giemsa staining, phalloidin staining, immunofluorescence and flow cytometry were used to identify the effects of 3,3ʹ-di-O-methylellagic acid 4ʹ-glucoside (DMAG), a natural ellagic acid derived from Sanguisorba officinalis L. (SOL) on megakaryocyte differentiation in HEL cells.
Results
DMAG significantly promoted megakaryocyte differentiation of HEL cells. DMAG administration accelerated platelet recovery and megakaryopoiesis, shortened tail bleeding time, strengthened platelet aggregation and adhesion in thrombocytopenia mice. Network pharmacology revealed that ITGA2B, ITGB3, VWF, PLEK, TLR2, BCL2, BCL2L1 and TNF were the core targets of DMAG. GO and KEGG pathway enrichment analyses suggested that the core targets of DMAG were enriched in PI3K–Akt signaling pathway, hematopoietic cell lineage, ECM-receptor interaction and platelet activation. Molecular docking simulation and molecular dynamics simulation further indicated that ITGA2B, ITGB3, PLEK and TLR2 displayed strong binding ability with DMAG. Finally, western blot analysis evidenced that DMAG up-regulated the expression of ITGA2B, ITGB3, VWF, p-Akt and PLEK.
Conclusion
DMAG plays a critical role in promoting megakaryocytes differentiation and platelets production and might be a promising medicine for the treatment of thrombocytopenia.

https://molmed.biomedcentral.com/articles/10.1186/s10020-021-00404-1
And from pomegranate:
“Effects of pomegranate juice and extract polyphenols on platelet function” (2009)
Several studies have shown that polyphenols reduce cardiovascular accidents in high-risk patients; in particular, the inhibition of platelet function may be responsible for part of this benefit. This research studied the antiplatelet effect of Wonderful variety pomegranate (Punica granatum) products, which contain primarily hydrolyzed tannins such as ellagitannins. We have investigated in vitro the effects of treatment with either pomegranate juice (PJ) or the polyphenol-rich extract from pomegranate fruit (POMx) on platelet aggregation, calcium mobilization, thromboxane A(2) production, and hydrogen peroxide formation, induced by collagen and arachidonic acid. PJ and POMx reduce all the platelet responses studied. POMx showed a stronger action in reducing platelet activation; moreover, POMx is active at the concentration that it is possible to obtain after polyphenol-rich food intake (2 microM). These results demonstrated that the cardiovascular health benefits of pomegranate may in part be related to the ability of polyphenols to inhibit platelet function. In fact, PJ and pomegranate extract have similar effects at concentrations expected for normal intake.
Further damage limitation is via anti-cancer therapeutics (teas come into this category too), but prevention is better than cure by limiting all exposure to RGD containing peptides.
If you know of any other potential therapeutics for thrombocytopenia please drop them in the comments? Thanks, DC.
Conclusion
The RBD region of SARS-CoV-2 and its GP120, GAG and Tat motifs is able to infect CD4+ T helper cells either directly or via receptors including CD26+ (DPP4), CCR4, CCR5, integrins & TMPRSS2.
One way another your CD4+ T cells will get infected at some point and depleted in a cumulative fashion by spike protein induced pathologies. Synergies between mechanisms may well also come into play.
Additional pathologies are induced by spike induced antibodies and anti-idiotype antibodies, in particular to platelets via binding to αVβ3 and α5β1 integrins, but αVβ3 interaction with the RGD containing peptides is also associated with AIDS-related pathologies including tumorigenesis, immune suppression, dementia, heart disease and lymphomas.
Spike protein expressing mRNA gene agents and adenoviral-vector vaccines also demonstrate a temporal association to a multi-fold increase in case reports of the above pathologies since their mass deployment from December 2020. As per my other Substacks, multiple pathways are implicated in this.

Disclaimer
This site is strictly an information website about pathophysiology, potential therapeutic agents and a review of associated research papers. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
References
Brunetti NS, Davanzo GG, Moraes D de, et al. SARS-CoV-2 Uses CD4 to Infect T Helper Lymphocytes. Published online January 11, 2023:2020.09.25.20200329. doi:10.1101/2020.09.25.20200329
https://www.medrxiv.org/content/10.1101/2020.09.25.20200329v2.full
Iyer SS, Cheng G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit Rev Immunol. 2012;32(1):23-63.
Pradhan P, Pandey AK, Mishra A, et al. Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag. Published online January 31, 2020:2020.01.30.927871. doi:10.1101/2020.01.30.927871
Abbott) labbott@thekmgroup co uk (Lauren. The full list of patients to be offered a Covid-19 booster jab by the NHS. Published 1691509173. Accessed August 9, 2023. https://www.msn.com/en-gb/health/medical/the-full-list-of-patients-to-be-offered-a-covid-19-booster-jab-by-the-nhs/ar-AA1eXLnD?ocid=entnewsntp&cvid=eed57ed5aea64287b4eec5c1fb67d0d0&ei=11
SARS-CoV-2 genome sequence prevalence and growth rate update: 2 August 2023. GOV.UK. Accessed August 9, 2023. https://www.gov.uk/government/publications/sars-cov-2-genome-sequence-prevalence-and-growth-rate/sars-cov-2-genome-sequence-prevalence-and-growth-rate-update-2-august-2023
UK releases recommendations for fall COVID boosters | CIDRAP. Published August 8, 2023. Accessed August 9, 2023. https://www.cidrap.umn.edu/covid-19/uk-releases-recommendations-fall-covid-boosters
Dianzani U, Bragardo M, Buonfiglio D, et al. Modulation of CD4 lateral interaction with lymphocyte surface molecules induced by HIV-1 gp120. Eur J Immunol. 1995;25(5):1306-1311. doi:10.1002/eji.1830250526
HUBACEK JA, DUSEK L, MAJEK O, et al. CCR5Δ32 Deletion as a Protective Factor in Czech First-Wave COVID-19 Subjects. Physiol Res. 2021;70(1):111-115. doi:10.33549/physiolres.934647
Morimoto C, Lord CI, Zhang C, Duke-Cohan JS, Letvin NL, Schlossman SF. Role of CD26/dipeptidyl peptidase IV in human immunodeficiency virus type 1 infection and apoptosis. Proc Natl Acad Sci U S A. 1994;91(21):9960-9964.
Barillari G, Sgadari C, Fiorelli V, et al. The Tat Protein of Human Immunodeficiency Virus Type-1 Promotes Vascular Cell Growth and Locomotion by Engaging the α5β1 and αvβ3 Integrins and by Mobilizing Sequestered Basic Fibroblast Growth Factor. Blood. 1999;94(2):663-672. doi:10.1182/blood.V94.2.663
Vankadari N, Wilce JA. Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 9(1):601-604. doi:10.1080/22221751.2020.1739565
Cameron K, Rozano L, Falasca M, Mancera RL. Does the SARS-CoV-2 Spike Protein Receptor Binding Domain Interact Effectively with the DPP4 (CD26) Receptor? A Molecular Docking Study. International Journal of Molecular Sciences. 2021;22(13):7001. doi:10.3390/ijms22137001
Sebastián-Martín A, Sánchez BG, Mora-Rodríguez JM, Bort A, Díaz-Laviada I. Role of Dipeptidyl Peptidase-4 (DPP4) on COVID-19 Physiopathology. Biomedicines. 2022;10(8):2026. doi:10.3390/biomedicines10082026
Makowski L, Olson-Sidford W, W. Weisel J. Biological and Clinical Consequences of Integrin Binding via a Rogue RGD Motif in the SARS CoV-2 Spike Protein. Viruses. 2021;13(2):146. doi:10.3390/v13020146
Magri F, Giordano S, Latini A, Muscianese M. New-onset cutaneous kaposi’s sarcoma following SARS-CoV-2 infection. Journal of Cosmetic Dermatology. 2021;20(12):3747-3750. doi:10.1111/jocd.14549
Ruzgas G, Eshan SH, Ramanathan S, et al. Immune Reconstitution Inflammatory Syndrome Presenting as Disseminated Kaposi Sarcoma. Cureus. 2023;15(2). doi:10.7759/cureus.34832
Zhou X, Zhu H, Luo C, et al. Targeting integrin α5β1 in urological tumors: opportunities and challenges. Front Oncol. 2023;13:1165073. doi:10.3389/fonc.2023.1165073
Krishn SR, Singh A, Bowler N, et al. Prostate cancer sheds the αvβ3 integrin in vivo through exosomes. Matrix Biol. 2019;77:41-57. doi:10.1016/j.matbio.2018.08.004
Casali BC, Gozzer LT, Baptista MP, Altei WF, Selistre-de-Araújo HS. The Effects of αvβ3 Integrin Blockage in Breast Tumor and Endothelial Cells under Hypoxia In Vitro. Int J Mol Sci. 2022;23(3):1745. doi:10.3390/ijms23031745
Kwakwa KA, Sterling JA. Integrin αvβ3 Signaling in Tumor-Induced Bone Disease. Cancers (Basel). 2017;9(7):84. doi:10.3390/cancers9070084
Kwakwa KA, Sterling JA. Integrin αvβ3 Signaling in Tumor-Induced Bone Disease. Cancers (Basel). 2017;9(7):84. doi:10.3390/cancers9070084
Berghoff AS, Kovanda AK, Melchardt T, et al. αvβ3, αvβ5 and αvβ6 integrins in brain metastases of lung cancer. Clin Exp Metastasis. 2014;31(7):841-851. doi:10.1007/s10585-014-9675-0
Petito E, Colonna E, Falcinelli E, et al. Anti-severe acute respiratory syndrome coronavirus-2 adenoviral-vector vaccines trigger subclinical antiplatelet autoimmunity and increase of soluble platelet activation markers. Br J Haematol. 2022;198(2):257-266. doi:10.1111/bjh.18245
Davenport R, Brohi K. Fibrinogen depletion in trauma: early, easy to estimate and central to trauma-induced coagulopathy. Crit Care. 2013;17(5):190. doi:10.1186/cc13021
Kurbel S. Jerne’s “immune network theory”, of interacting anti-idiotypic antibodies applied to immune responses during COVID-19 infection and after COVID-19 vaccination. Bioessays. Published online June 9, 2023:e2300071. doi:10.1002/bies.202300071
DPP-4 Inhibitors (Gliptins) - Suitability, Benefits & Side Effects. Accessed August 12, 2023. https://www.diabetes.co.uk/diabetes-medication/dpp-4-inhibitors.html
Mohammadinejad A, Mohajeri T, Aleyaghoob G, Heidarian F, Kazemi Oskuee R. Ellagic acid as a potent anticancer drug: A comprehensive review on in vitro, in vivo, in silico, and drug delivery studies. Biotechnology and Applied Biochemistry. 2022;69(6):2323-2356. doi:10.1002/bab.2288
Yousuf M, Shamsi A, Khan P, et al. Ellagic Acid Controls Cell Proliferation and Induces Apoptosis in Breast Cancer Cells via Inhibition of Cyclin-Dependent Kinase 6. Int J Mol Sci. 2020;21(10):3526. doi:10.3390/ijms21103526
“my aim is to present the evidence which quite often is scant in number or hidden away on journal sites or even retracted if it doesn’t fit the political (or shareholder focused) narrative of the day.”
Which is why I try to read every one of your posts- no matter how dense the material is. You have a lovely writing style which is easy to follow- much appreciated! And your discoveries are always on target, you’ve been mentioning the HIV insert for ages now, and it’s only now I hear other voices agreeing and chiming in.
There are many actually ! Olive leaf extract is kind of strong and Dr.grouf/hachi recommended not to take straight for more than 7 days unless infected ...( full spectrum one is better for regular use I suppose ) , so presently am regular on pomegranate, papaya leaf rarely and E sometimes.. ..
Current strains are causing brain fog , headache and fatigue and overall weakness maybe more than previous ones ( personal experience) ...