25th June ‘22: NAC as a therapeutic; gp120 impairs tubulin function by binding to tubulin isoforms and inhibiting proper assembly of microtubules.
23rd July ‘22: Disruption of the kynurenine pathway; tunneling nanotubes provide a route for SARS-CoV-2 spreading.
3rd January ‘23: Hyperlinked contents page; effects of gp120 on fast axonal transport (FAT).
16th May ‘23: Behavioral Evidence for a Tau and HIV-gp120 Interaction
8th June ‘23: Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies.
Gp120 binding to LPS’s, proinflammatory cytokine response, immune exhaustion and neurotoxicity - lay off the meth!
15th October ‘23: New pre-proof published and how it further supports the gp120 hypothesis: “Brain fog in long COVID: A glutamatergic hypothesis with astrocyte dysfunction accounting for brain PET glucose hypometabolism” (13th Oct. ‘23)
Glioblastoma cases and gp120.
Any extracts used in the following article are for non commercial research and educational purposes only and may be subject to copyright from their respective owners.
Multiple factors appear to contribute to the SARS-CoV-19 - spike protein induced pathophysiology known as “brain fog”. These can include microthrombi, stroke, misfolded proteins, miRNA charged exosome induced microglial inflammation, linoleic acid disruption, B12 depletion, NAD+ depletion, and something else.
These are all currently hypotheses pending further research, and each may be responsible to a lesser or greater extent in different patient groups.
A key factor appears to be inflammatory cytokines:
Scientists Think They've Figured Out What's Triggering Brain Fog in COVID-19 Patients (2021)
Not long after the first wave of COVID-19 infections hit, doctors all around the world began to notice something strange – a host of lingering effects persisting in patients, long after they appeared to have otherwise recovered from the virus.
These unusual neurological symptoms – encompassing fatigue, memory loss, confusion, and other abnormalities – are sometimes known as 'brain fog' or 'COVID brain', and new research may have identified an underlying cause of the condition.
"We were initially approached by our colleagues in critical care medicine who had observed severe delirium in many patients who were hospitalised with COVID-19," says neuro-oncologist Jessica Wilcox from the Memorial Sloan Kettering Cancer Centre (MSK) in New York.
"That meeting turned into a tremendous collaboration between neurology, critical care, microbiology, and neuroradiology to learn what was going on and to see how we could better help our patients."
As part of the new study, Wilcox and fellow researchers screened the cerebrospinal fluid of 18 cancer patients who were experiencing neurological dysfunction (aka encephalopathy) after having been infected with the SARS-CoV-2 virus.
Initially, it was suspected that an ongoing viral infection might be the cause of their brain fog symptoms, but microbiological analysis of fluid taken in spinal taps did not reveal any sign of the virus, suggesting the patients had recovered from COVID-19.
Nonetheless, the search did turn up an important clue as to what was going on.
"We found that these patients had persistent inflammation and high levels of cytokines in their cerebrospinal fluid, which explained the symptoms they were having," explains MSK researcher and co-first author of the study, Jan Remsik.
Cytokines are a broad category of proteins that are involved with signalling in the immune system.
In some cases of coronavirus, an over-production of these molecules results in what's known as a cytokine storm, which can cause excessive inflammation and is potentially deadly.
A similar phenomenon showing high levels of inflammatory cytokines is sometimes seen as a side effect of chimeric antibody receptor (CAR) T cell therapy, an immunotherapy treatment, which can also produce confusion, delirium, and other neurological effects that bear a resemblance to COVID brain fog.
The thinking is that the flood of these inflammatory chemicals in the immune system seeps into the brain, producing symptoms of encephalopathy as seen in patients.
While this is the largest study to date to demonstrate this potential link between COVID-19 and post-infection neurological effects, we'll need a lot more data to untangle this association.
That said, the findings here suggest anti-inflammatory drugs might be helpful in mitigating brain fog in patients, and could highlight new directions in terms of diagnosing this strange, lingering malaise.
"We used to think that the nervous system was an immune-privileged organ, meaning that it didn't have any kind of relationship at all with the immune system," MSK neuro-oncologist Adrienne Boire explains.
"But the more we look, the more we find connections between the two."
Depletion of nicotinamide adenine dinucleotide (NAD+) is suspected, certainly in severe infections:
NAD+ in Brain Aging and Neurodegenerative Disorders (2020)
Abstract
NAD+ is a pivotal metabolite involved in cellular bioenergetics, genomic stability, mitochondrial homeostasis, adaptive stress responses, and cell survival. Multiple NAD+-dependent enzymes are involved in synaptic plasticity and neuronal stress resistance. Here, we review emerging findings that reveal key roles for NAD+ and related metabolites in the adaptation of neurons to a wide range of physiological stressors and in counteracting processes in neurodegenerative diseases, such as those occurring in Alzheimer’s, Parkinson’s, and Huntington diseases, and amyotrophic lateral sclerosis. Advances in understanding the molecular and cellular mechanisms of NAD+-based neuronal resilience will lead to novel approaches for facilitating healthy brain aging and for the treatment of a range of neurological disorders.
Nicotinamide adenine dinucleotide (NAD+) is a fundamental molecule in health and disease, as it is central to several cellular bioenergetic functions. NAD+ is synthesized via three major pathways, including de novo biosynthesis, the Preiss-Handler pathway, and the salvage pathway (Figure 1).
Severe viral infections, including COVID-19, can trigger the inflammatory pathways & depletion in a feedback loop:
NAD+ in COVID-19 and viral infections (2022)
Abstract
NAD+, as an emerging regulator of immune responses during viral infections, may be a promising therapeutic target for coronavirus disease 2019 (COVID-19). In this Opinion, we suggest that interventions that boost NAD+ levels might promote antiviral defense and suppress uncontrolled inflammation. We discuss the association between low NAD+ concentrations and risk factors for poor COVID-19 outcomes, including aging and common comorbidities. Mechanistically, we outline how viral infections can further deplete NAD+ and its roles in antiviral defense and inflammation. We also describe how coronaviruses can subvert NAD+-mediated actions via genes that remove NAD+ modifications and activate the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome. Finally, we explore ongoing approaches to boost NAD+ concentrations in the clinic to putatively increase antiviral responses while curtailing hyperinflammation.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and other viral infections render the body a battlefield, requiring mobilization of vast resources to mount an effective defense. These defenses can backfire when uncontrolled, resulting in deadly cytokine storms (see Glossary). A key to effective interventions is to trigger robust antiviral defenses but with checked inflammation. Therefore, molecules that suppress both viral replication and inflammation may be particularly important for fighting severe COVID-19 [1]. A growing body of evidence shows that the metabolite NAD+ is a mediator of both antiviral and anti-inflammatory mechanisms. Based on this evidence, we posit that therapies that boost NAD+ concentrations might play a role in preventing and treating severe COVID-19 and other viral infections.
First described as a yeast fermentation factor over a century ago, today, NAD+ has risen to prominence as a regulator of healthy aging. Low levels of NAD+ in tissues and organs are associated with aging, metabolic syndrome, and inflammation, while dietary interventions that slow age-related diseases increase NAD+ concentrations [2]. Here, we review the epidemiological and mechanistic data supporting a role for NAD+ in modulating the outcomes of viral infections, with a focus on SARS-CoV and SARS-CoV-2. We also explore ongoing approaches to boost NAD+ levels for therapeutic benefit in the clinic.
Mechanisms of NAD+ in viral infections
Viral infections can deplete NAD+ concentrations
Not only are low NAD+ concentrations associated with risk factors for poor COVID-19 outcomes, but certain viral infections can further deplete NAD+ in infected cells. For example, lower NAD+ concentrations have been reported in human peripheral blood leukocytes infected with HIV-1 in vitro [35], human fibroblasts infected with herpes simplex virus 1 (HSV-1) [36], and in the skeletal muscle of individuals coinfected with HIV-1 and hepatitis C virus [37]. These effects are presumably due to the induction of NAD+-consuming enzymes such as CD38 and PARPs [17]. For instance, CD8+ T lymphocytes expressing CD38 have been proposed as a marker for HIV-1-mediated disease progression [38]. The decline of NAD+ concentrations in human fibroblasts induced by HSV-1 infection is associated with increased protein poly(ADP-ribosyl)ation, and can be blocked by pharmacological inhibition of PARP1/PARP2 [36].
Similar depletion of NAD+ has been reported with coronavirus infections. Specifically, blood from severe COVID-19 patients contains lower amounts of the NAD+ precursor NMN compared with blood from healthy individuals [39]. Moreover, expression changes in genes involved in NAD+ synthesis and utilization such as Nampt, Parp9, Parp12, and Parp14 have been observed in epithelial cell lines and enterocyte organoids infected with SARS-CoV-2 compared with mock infection [40]. Similar NAD+-related gene expression changes were also reported in the lung tissue of a deceased COVID-19 patient and in the bronchoalveolar lavage fluid of SARS-CoV-2-infected individuals relative to healthy controls [40]. Furthermore, infection of a coronavirus, mouse hepatitis virus (MHV), in mouse bone-marrow-derived macrophages (BMDMs) induced NAD+ depletion [40] as well as increased gene expression of many PARPs including Parp7, Parp9, Parp10, Parp11, Parp12, Parp13, and Parp14 relative to mock infection [41]. These observations suggest that NAD+ metabolic pathways are under increased demand during SARS-CoV-2 infection, and highlights the potential relevance of NAD+ in modulating COVID-19 disease outcomes.
NAD+-consuming enzymes in antiviral mechanisms
NAD+ harbors an important role in fueling the activity of enzymes that regulate mammalian immune responses [42,43]. Classically, NAD+ participates in redox processes, but it also participates in non‐redox reactions in which it is hydrolyzed, and it non‐enzymatically regulates protein–protein interactions [16] (Figure 1). In these latter functions, NAD+ acts as a signaling molecule, serving as a marker of energy availability and directing a cell to respond to metabolic changes via the action of NAD+-utilizing enzymes [44].
PARPs and sirtuins are two NAD+-dependent enzyme families that participate in immune responses [42,43]. By adding or removing post-translational modifications on key proteins such as nuclear factor kappa B (NF-κB), they can coordinate the intensity of inflammatory and immune responses [42]. This places NAD+ in an important position for both promoting strong immune responses to pathogens, and for keeping those responses in check.
PARP1, the pre-eminent member of the PARP family, is a potent coactivator of the proinflammatory transcription factor NF-κB, and therefore participates in initiating specific immune responses [42]. However, this is a double-edged sword, as PARP1 may also increase the severity of cytokine storms as it regulates the expression of many NF-κB-dependent cytokines and chemokines [42]. Indeed, inhibiting or deleting PARP1 has ameliorated the severity of symptoms in several inflammatory disease rodent models including asthma and colitis [45., 46., 47., 48., 49., 50., 51.]. For example, PARP1 inhibition (pharmacological or genetic) has prevented ovalbumin-induced lung inflammation in mice [45] and in a guinea pig model of asthma [46]. Treatment with PARP inhibitors has attenuated inflammation associated with colitis seen in interleukin-10 (IL-10) deficient (Il10-/-) mice [47], as well as in trinitrobenzene sulfonic acid-treated rats [49]. Furthermore, PARP1 knockout (KO) (Parp1-/-) mice are protected from dextran sulfate sodium-induced colitis compared with wild-type mice [48].
Several PARPs harbor potent antiviral functions [42,52., 53., 54., 55., 56., 57., 58., 59.]. Indeed, PARP13 is a powerful antiviral factor that recognizes various viruses from several families, including Retroviridae, Filoviridae, Alphaviridae, and Hepadnaviridae [53., 54., 55., 56.]. It binds to specific sequences of viral RNAs during infection and mediates their degradation via the cellular mRNA decay machinery; however, these functions are not dependent on PARP-mediated ADP-ribosylation [54., 55., 56.]. Expression of PARP7, PARP10, and the long isoform of PARP12 (PARP12L) efficiently inhibits cellular translation and the replication of Venezuelan equine encephalitis virus and other alphaviruses in vertebrate cells [52., 59.]. These effects of PARP12L are dependent on its catalytic activity. Moreover, PARP9 and PARP14 are also upregulated in macrophages stimulated by interferon (IFN)-γ and have opposing roles in macrophage activation [58]. PARP9 activates IFNγ–STAT1 signaling and induces proinflammatory activation while PARP14 ADP-ribosylation reduces STAT1 phosphorylation in IFNγ-treated human macrophages [58]. , Additionally, the nucleocapsid proteins of several coronaviruses, including SARS-CoV, MERS-CoV, and MHV, are ADP-ribosylated in infected cells, presumably by PARPs, which may indicate a common use of this pathway among viruses. However, the functional consequences of such ADP‐ribosylation remain to be investigated [57]. We argue that since PARP enzymatic activity requires NAD+[44], maintaining a sufficient NAD+ concentration may be crucial for achieving PARP-related antiviral mechanisms.
Sirtuins also play a role in antiviral defenses [43,60., 61., 62., 63., 64.]. Indeed, sirtuin 1 (SIRT1) KO or inhibition promotes the lifecycle and replication of vesicular stomatitis virus in mouse embryonic fibroblasts (MEFs) and Kaposi’s sarcoma-associated herpesvirus in human lymphoma cell lines [61,62]. Disruption of SIRT1 also increases HPV16 E1–E2 replication [60]. Moreover, knockdown via siRNA of each of the seven sirtuins in human fibroblast cells promoted the growth of a diverse set of human viruses after infection, including human cytomegalovirus (CMV), HSV1, adenovirus type 5, and influenza virus (H1N1) [63]. Furthermore, SIRT1-activating drugs such as resveratrol and CAY10602 have inhibited the replication of these viruses [63]. SIRT6 promotes tumor necrosis factor (TNF)α secretion, as evidenced from the suppression of TNFα release from SIRT6 KO MEFs, whereby TNFα secretion would be expected to promote the eradication of pathogens [64]. Indeed, pharmacological inhibition and siRNA knockdown of SIRT6 and NAMPT, an NAD+-synthetic enzyme (Figure 1), in mouse fibroblasts promoted CMV replication [65].
Other NAD+-utilizing enzymes that also play roles during immune responses include CD38 [66], BST1 [2], and SARM1 [43]. Indeed, CD38 and BST1 are highly expressed on the surface of macrophages and lymphocytes and produce extracellular cyclic(ADP-ribose), a calcium-mobilizing second messenger that is important for immune cell activation [2,66,67]. SARM1, another potent NADase, contains a Toll/IL-1 receptor domain, which might elicit neuroprotective innate immune responses, as suggested from mouse models of neurodegeneration [68].
Due to the GP120 & GAG inserts I thought I would do a cross reference with HIV.
Note the doom loop he also refers to and the need to break the cycle with therapeutics:
Clinical Relevance of Kynurenine Pathway in HIV/AIDS: An Immune Checkpoint at the Crossroads of Metabolism and Inflammation
Jean-Pierre Routy et al. AIDS Rev. 2015 Apr-Jun.
Abstract
Tryptophan degradation along the kynurenine pathway is associated with a wide variety of pathophysiological processes, of which tumor tolerance and immune dysfunction in several chronic viral infections including HIV are well known. The kynurenine pathway is at the crossroads of metabolism and immunity and plays an important role in inflammation while also playing an opposing role in the control of acute and chronic infections. In this review we have summarized findings from recent studies reporting modulation of tryptophan degrading the kynurenine pathway in the context of HIV infection. This immuno-metabolic pathway is modulated by three distinct inducible enzymes: indoleamine 2,3-dioxygenase 1 and 2 and tryptophan 2,3-dioxygenase. Increased expression of these enzymes by antigen-presenting cells leads to local or systemic tryptophan depletion, resulting in a mechanism of defense against certain microorganisms. Conversely, it can also lead to immunosuppression by antigen-specific T-cell exhaustion and recruitment of T regulatory cells. Recently, among these enzymes, indoleamine 2,3-dioxygenase 1 has been recognized to be an immune response checkpoint that plays an important role in HIV immune dysfunction, even in the context of antiretroviral therapy. In addition to the activation of the kynurenine pathway by HIV proteins Tat and Nef, the tryptophan-degrading bacteria present in the intestinal flora have been associated with dysfunction of gut mucosal CD4 Th17/Th22 cells, leading to microbial translocation and creating a systemic kynurenine pathway activation cycle. This self-sustaining feedback loop has deleterious effects on disease progression and on neurocognitive impairment in HIV-infected patients. Therapy designed to break the vicious cycle of induced tryptophan degradation is warranted to revert immune exhaustion in HIV-infected persons.
Whilst virus infections can deplete NAD+ and disrupt the KP as above, it would appear that this may not be the whole story, to explain all the symptomology.
A mild infection may be sufficient to induce it, but is there anything else that is also known to cause the same symptomology and may be implicated too?
Again we need to look to research into HIV for clues.
HIV Neurotoxicity: Potential Therapeutic Interventions (2006)
Abstract
Individuals suffering from human immunodeficiency virus type 1 (HIV-1) infection suffer from a wide range of neurological deficits. The most pronounced are the motor and cognitive deficits observed in many patients in the latter stages of HIV infection. Gross postmortem inspection shows cortical atrophy and widespread neuronal loss. One of the more debilitating of the HIV-related syndromes is AIDS-related dementia, or HAD. Complete understanding of HIV neurotoxicity has been elusive. Both direct and indirect toxic mechanisms have been implicated in the neurotoxicity of the HIV proteins, Tat and gp120. The glutamatergic system, nitric oxide, calcium, oxidative stress, apoptosis, and microglia have all been implicated in the pathogenesis of HIV-related neuronal degeneration. The aim of this review is to summarize the most recent work and provide an overview to the current theories of HIV-related neurotoxicity and potential avenues of therapeutic interventions to prevent the neuronal loss and motor/cognitive deficits previously described.
GENERAL MECHANISMS OF HIV NEUROTOXICITY
Just over a decade ago, the first reports of HIV-related neurotoxicity were published. Two proteins associated with the AIDS virus, gp120 (a coat glycoprotein) and Tat (transactivation) have been shown to be neurotoxic. The HIV-associated protein gp120 was shown to be neurotoxic to cultured dopamine neurons [1]. Exposure to gp120 for 3 days reduced the ability of neurons to transport dopamine and decreased the size of the dendritic tree.
We start by looking for envelope glycoprotein GP120 in COVID-19 as we know from the above that this is neurotoxic and also involved with brain fog symptoms, in as little as 3 days.
Neurotoxicity, a key to pathology at low molar concentrations and a major contributor to brain fog mediated in part by NAD+ depletion & inflammatory cytokines?
But GP120 is in HIV, so I must be on the wrong track, end of post, thanks for tuning in…
Envelope glycoprotein GP120 (or gp120) is a glycoprotein exposed on the surface of the HIV envelope. It was discovered by Professors Tun-Hou Lee and Myron "Max" Essex of the Harvard School of Public Health in 1988.[1] The 120 in its name comes from its molecular weight of 120 kDa. Gp120 is essential for virus entry into cells as it plays a vital role in attachment to specific cell surface receptors.
Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag (2020)
It was later retracted, but as fraud or experimental techniques were not seriously in question it appears to be more at the request of Corporate.
Abstract
We are currently witnessing a major epidemic caused by the 2019 novel coronavirus (2019-nCoV). The evolution of 2019-nCoV remains elusive. We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV-1 gp120 or HIV-1 Gag. Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature. This work provides yet unknown insights on 2019-nCoV and sheds light on the evolution and pathogenicity of this virus with important implications for diagnosis of this virus.
FIGURE 3.Modelled homo-trimer spike glycoprotein of 2019-nCoV virus. The inserts from HIV envelop protein are shown with colored beads, present at the binding site of the protein.
The first 3 inserts (insert 1,2 and 3) aligned to short segments of amino acid residues in HIV-1 gp120. The insert 4 aligned to HIV-1 Gag. The insert 1 (6 amino acid residues) and insert 2 (6 amino acid residues) in the spike glycoprotein of 2019-nCoV are 100% identical to the residues mapped to HIV-1 gp120. The insert 3 (12 amino acid residues) in 2019-nCoV maps to HIV-1 gp120 with gaps [see Table 1]. The insert 4 (8 amino acid residues) maps to HIV-1 Gag with gaps.
Although, the 4 inserts represent discontiguous short stretches of amino acids in spike glycoprotein of 2019-nCoV, the fact that all three of them share amino acid identity or similarity with HIV-1 gp120 and HIV-1 Gag (among all annotated virus proteins) suggests that this is not a random fortuitous finding. In other words, one may sporadically expect a fortuitous match for a stretch of 6-12 contiguous amino acid residues in an unrelated protein. However, it is unlikely that all 4 inserts in the 2019-nCoV spike glycoprotein fortuitously match with 2 key structural proteins of an unrelated virus (HIV-1).
The amino acid residues of inserts 1, 2 and 3 of 2019-nCoV spike glycoprotein that mapped to HIV-1 were a part of the V4, V5 and V1 domains respectively in gp120 [Table 1]. Since the 2019-nCoV inserts mapped to variable regions of HIV-1, they were not ubiquitous in HIV-1 gp120, but were limited to selected sequences of HIV-1 [refer S.File1] primarily from Asia and Africa.
The HIV-1 Gag protein enables interaction of virus with negatively charged host surface (Murakami, 2008) and a high positive charge on the Gag protein is a key feature for the host-virus interaction. On analyzing the pI values for each of the 4 inserts in 2019-nCoV and the corresponding stretches of amino acid residues from HIV-1 proteins we found that a) the pI values were very similar for each pair analyzed b) most of these pI values were 10±2 [Refer Table 1]. Of note, despite the gaps in inserts 3 and 4 the pI values were comparable. This uniformity in the pI values for all the 4 inserts merits further investigation.
As none of these 4 inserts are present in any other coronavirus, the genomic region encoding these inserts represent ideal candidates for designing primers that can distinguish 2019-nCoV from other coronaviruses.
Unfortunately, retracting the paper without evidence of fraud doesn’t make the variable loop inserts and their pathologies go away:
Glycan reactive anti-HIV-1 antibodies bind the SARS-CoV-2 spike protein but do not block viral entry (2021)
We have identified a novel set of cross-reactive interactions between the SARS-CoV-2 spike and broadly neutralizing anti-HIV antibodies. A recent study15 has demonstrated such cross-reactivity using Fab-dimerized antibodies such as 2G12, which target high mannose epitopes with gp120, and here we have extended these results to include anti-HIV antibodies which recognize gp120 at epitopes involving both peptide and glycans. Using a combination of immunoblotting, ELISA, and immunoprecipitation experiments, we demonstrate PGT128 and PGT126 bind both ectodomain and full-length SARS-CoV-2 S constructs, under denaturing and native conditions, in a glycan dependent manner.
And this paper, which demonstrated how gp120 can bind to LPS’s and exacerbate the pro-inflammatory cytokine response, contributing to T-cell exhaustion:
Interaction of the HIV-1 gp120 Viral Protein V3 Loop with Bacterial Lipopolysaccharide (2011)
HIV-1 represents an elusive target for therapeutic compounds due to its high rate of mutation. Targeting structural patterns instead of a constantly changing specific three-dimensional structure may represent an approach that is less sensitive to viral mutations. The V3 loop of gp120 of HIV-1, which is responsible for binding of viral gp120 to CCR5 or CXCR4 coreceptors, has already been identified as an effective target for the inhibition of viral entry. The peptide derived from the V3 loop of gp120 specifically interacts with the lipid A moiety of LPS, as does the full gp120 protein. NMR analysis of V3 in complex with LPS shows formation of an amphipathic turn. The interaction between LPS and V3 relies on the structural pattern, comprising a combination of hydrophobic and charge interactions, similar to the interaction between antimicrobial peptides and LPS. LPS inhibited binding of gp120 to the surface of target T cells. Nonendotoxic LPS antagonists inhibited viral infection, demonstrating the possibility for the development of an inhibitor of HIV-1 attachment to T cells based on the recognition of a conserved structural pattern.
A common structural motif of the V3 loop in complex with neutralizing antibodies, based on experimental results and computer modeling, indicates that V3 comprises GPG(R/K/Q) sequence at the center of the loop, two extended regions flanking the central crest, and a helical region at the C-terminal segment (16, 17, 18, 19). Four conserved structural elements in the V3 loop and two additional variable regions have been revealed based on cross-clade neutralizing antibodies and V3 peptides derived from various HIV-1 strains (20). Because the V3 peptide is unstructured when free in solution, it probably binds to the antibodies by an induced fit mechanism and adopts a conformation that is complementary to the antibody paratope.
HIV-1 infection results in chronic immune activation, contributing to immune dysfunction (21). Increased levels of bacterial lipopolysaccharide (LPS) and components of the LPS-activating complex have been detected in the plasma of chronically infected HIV-1-positive individuals (22). LPS is a major component of the outer cell membrane of Gram-negative bacteria. It is an amphipathic molecule, consisting of a variable hydrophilic segment comprising the polysaccharide core and O-antigen and a more conserved hydrophobic region known as lipid A. LPS is the principal stimulator of the innate immune response to Gram-negative bacteria. It is recognized by a complex cascade of extracellular “pattern recognition receptors,” which recognize the conserved motif of lipid A and present LPS to the lipid A-binding protein, MD-2, associated with the transmembrane receptor TLR4 (23). Recognition of various forms of LPS from different strains of Gram-negative bacteria is accomplished through the lipid A moiety, which represents the pathogen-associated molecular pattern, triggering the signaling cascade, which results in a release of pro-inflammatory mediators, such as cytokines, chemokines, and others (24). The conserved lipid A moiety is the minimal structural fragment of LPS that triggers the cellular response (25). Although there are structural differences among key extracellular LPS-sensing and -relaying receptors, lipopolysaccharide-binding protein, CD14, and MD-2, the lipid A recognition motif can be reduced to the arrangement of negatively charged groups at the defined distance with a large adjacent hydrophobic patch (24). There are many studies on the role of LPS in HIV-1 replication or inhibition. Contradictory studies may be a consequence of the activation immune system receptors, which may result in the transcriptional activation or repression of gp120 coreceptors. For example, although it has been shown that LPS inhibits HIV-1 replication in macrophages (26), several studies have also shown that LPS enhances HIV-1 replication in monocytoid cell lines (27). As we show in this study, it may also impart its effect through direct binding to gp120.
Our research was initiated by the finding that a peptide from the V3 loop of gp120 exerted antimicrobial activity against Gram-negative bacteria (28). We demonstrate that gp120 and the 15-residue V3 peptide (29) directly and specifically interact with different chemotypes of LPS. We determined the conformation of the V3 peptide in complex with LPS using high resolution NMR and docking methods. The specific binding of V3 to LPS was confirmed by fluorescence spectroscopy, by a binding assay, and by the neutralization of activation of a monocytoid cell line Mono Mac 6. We demonstrated that LPS inhibited binding of gp120 to the receptors on a target T cell line H9. Nonendotoxic LPS antagonists (tetraacylated MLK986 LPS and lipid IVa) inhibited infection by an HIV-1 pseudovirus in the U87.CD4.CCR5 cell line, indicating that nonendotoxic compounds could be potentially used in an anti-HIV therapy.
Advice is to lay off the meth if you have circulating gp120:
Methamphetamine and HIV-1 gp120 Effects on Lipopolysaccharide Stimulated Matrix Metalloproteinase-9 Production by Human Monocyte-Derived Macrophages (2011)
Abstract
Monocytes/macrophages are a primary source of human immunodeficiency virus (HIV-1) in the central nervous system (CNS). Macrophages infected with HIV-1 produce a plethora of factors, including matrix metalloproteinase-9 (MMP-9) that may contribute to the development of HIV-1-associated neurocognitive disorders (HAND). MMP-9 plays a pivotal role in the turnover of the extracellular matrix (ECM) and functions to remodel cellular architecture. We have investigated the role of methamphetamine and HIV-1 gp120 in the regulation of lipopolysaccaride (LPS) induced-MMP-9 production in monocyte-derived macrophages (MDM). Here, we show that LPS-induced MMP-9 gene expression and protein secretion are potentiated by incubation with methamphetamine alone and gp120 alone. Further, concomitant incubation with gp120 and methamphetamine potentiated LPS-induced MMP-9 expression and biological activity in MDM. Collectively methamphetamine and gp120 effects on MMPs may modulate remodeling of the extracellular environment enhancing migration of monocytes/macrophages to the CNS.
Matrix metalloproteinases (MMPs), a family of zinc- and calcium-dependent endopeptidases, catalyze the proteolysis of the extracellular matrix (ECM) (Kaczmarek et al., 2002; Dzwonek et al., 2004). MMPs are produced by numerous cell types including monocytes, macrophages, lymphocytes, neutrophils, and eosinophils (Visse and Nagasse, 2003). MMPs are synthesized as inactive precursors or proenzymes and are activated following proteolytic removal of the propeptide domain.
MMPs mediate many normal and pathologic biological processes, including cell migration, invasion, proliferation, angiogenesis, embryogenesis and tissue remodeling (Mannello and Gazzanelli, 2001; Van den Steen et al., 2002). MMPs degrade constituents of the ECM contributing to blood-brain barrier (BBB) leakage and infiltration by activated or infected immune cells (Mun-Bryce et al., 1998; Vos et al., 2000; Van den Steen et al., 2002). Tissue inhibitors of metalloproteinase (TIMPs) regulate the proteolytic activity of MMPs (Mannello and Gazzanelli 2001). Collectively, MMPs and TIMPs modulate remodeling of the extracellular environment.
Figure 1. The effect of methamphetamine and/or gp120 on LPS-stimulated MMP-9 gene expression from MDM. MDM were incubated with methamphetamine alone (5 μM), gp120 alone (50 ng/ml) or concomitantly for 24 h, MDM were washed then stimulated with LPS (100 ng/ml) for 12 h. RNA was isolated and gene expression for MMP-9 was determined using Q-PCR. Meth = methamphetamine. Statistical significance was calculated using ANOVA followed by Bonferroni post-hoc test, *compared to control (p < 0.005); †compared to LPS alone (p < 0.001). Data represent the mean ± standard deviation, n = 4.
This study sought to investigate the effect that the addictive drug methamphetamine and the HIV-1 viral protein gp120 have on the production of MMP-9 by LPS stimulated macrophage. MMP-9 production has been associated with disruption of the BBB which may increase the risk for development of HAND (Power et al., 1993; Dallasta et al., 1999; Webster and Crowe, 2006; Louboutin et al., 2010).
Aberrant immune activation associated with HIV-1 disease has been correlated with microbial or microbial by-product translocation into the peripheral blood as a consequence of gastrointestinal tract impairment (Brenchley and Douek, 2008). LPS, a component of the outer membrane of gram-negative bacteria, is an endotoxin that elicits a strong immune response. Brenchley et al. found circulating levels of plasma LPS to be significantly increased in chonically infected HIV-1 patients (Brenchley et al., 2006). Antiretroviral therapy decreased plasma LPS levels (Brenchley et al., 2006; Baroncelli et al., 2009).
Moreover, studies demonstrated an association between a reduction in the levels of plasma LPS and CD4 T-cell reconstitution following antiretroviral therapy (Jiang et al., 2009; Ciccone et al., 2010). Furthermore, subjects with acute and chonic HIV-1 infection had higher levels of plasma soluble CD14 (sCD14, marker of macrophage activation) than HIV-seronegative subjects suggesting a role of activated monocyte/macrophage in the progression of HIV-1 disease (Brenchley et al., 2006).
The incidence and severity of HAND are exacerbated by drugs of abuse (Nath et al., 2001; Ferris et al., 2009). Addictive drug use is associated with an increase in bacterial infections, i.e. increased LPS production (Friedman et al., 2003). Ancuta et al, demonstrated that LPS levels were higher in AIDS patients with IV heroin or ethanol abuse and were lower in patients on HAART, compared to control (Ancuta et al., 2008). This study also found high plasma LPS and LPS-binding protein (LBP) levels, together with low endotoxin core antibody (EndoCAb) levels, were associated with increased soluble markers of monocyte/macrophage activation and HAND in drug users (Ancuta et al., 2008). These authors suggest a role for drug abuse as a cofactor that produces elevated LPS levels in triggering monocyte/macrophage activation thereby contributing to the incidence and severity of HAND.
Research strongly suggests COVID-19 virus enters the brain (2020)
A study published in Nature Neuroscience shows how spike protein crosses the blood-brain barrier.
Banks said the S1 protein in SARS-CoV2 and the gp 120 protein in HIV-1 function similarly. They are glycoproteins – proteins that have a lot of sugars on them, hallmarks of proteins that bind to other receptors. Both these proteins function as the arms and hand for their viruses by grabbing onto other receptors. Both cross the blood-brain barrier and S1, like gp120, is likely toxic to brain tissues.
“It was like déjà vu,” said Banks, who has done extensive work on HIV-1, gp120, and the blood-brain barrier.
Ok ok we get it, it's either GP120 or a very close match with similar pathophysiology. It's like we are dealing with artificial insertion of the sequences, gain of function work, bioweapon research? Nah, must be due to bats or pangolins.
Moving on...So what happens next, once it contacts a neuron? The quick answer is that it may inhibit neurotransmission or even lead to the death of your brain cell, your neuron, by apoptosis, symptomatic to the recipient as “brain fog” if this is happening to enough neurons.
Key point is that the presence of live viral mRNA is not a prerequisite here, just the glycoprotein alone is sufficient to cause neurological effects.
Read on…
Potassium gating
Both NAD+ depletion and GP120 can affect potassium gating:
NAD+ activates KNa channels in dorsal root ganglion neurons (2009)
Abstract
Although sodium-activated potassium channels (KNa) have been suggested to shape various firing patterns in neurons, including action potential repolarization, their requirement for high concentrations of Na+ to gate conflicts with this view. We characterized KNa channels in adult rat dorsal root ganglion (DRG) neurons. Using immunohistochemistry, we found ubiquitous expression of the Slack KNa channel subunit in small-, medium-, and large-diameter DRG neurons. Basal KNa channel activity could be recorded from cell-attached patches of acutely dissociated neurons bathed in physiological saline, and yet in excised inside-out membrane patches, the Na+ EC50 for KNa channels was typically high, approximately 50 mM. In some cases, however, KNa channel activity remained considerable after initial patch excision but decreased rapidly over time. Channel activity was restored in patches with high Na+. The channel rundown after initial excision suggested that modulation of channels might be occurring through a diffusible cytoplasmic factor. Sequence analysis indicated that the Slack channel contains a putative nicotinamide adenine dinucleotide (NAD+)-binding site; accordingly, we examined the modulation of native KNa and Slack channels by NAD+. In inside-out-excised neuronal patch recordings, we found a decrease in the Na+ EC50 for KNa channels from approximately 50 to approximately 20 mM when NAD+ was included in the perfusate. NAD+ also potentiated recombinant Slack channel activity. NAD+ modulation may allow KNa channels to operate under physiologically relevant levels of intracellular Na+ and hence provides an explanation as to how KNa channel can control normal neuronal excitability.
Target of HIV-1 Envelope Glycoprotein gp120–Induced Hippocampal Neuron Damage: Role of Voltage-Gated K+ Channel Kv2.1 (2015)
Abstract
“Human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein 120 (gp120) has been reported to be toxic to the hippocampal neurons, and to be involved in the pathogenesis of HIV-1-associated neurocognitive disorders (HAND). Accumulating evidence has demonstrated that voltage-gated potassium (Kv) channels, especially the outward delayed-rectifier K+ (Ik) channels, play a critical role in gp120-induced cortical neuronal death in vitro. However, the potential mechanisms underlying the hippocampal neuronal injury resulted from gp120-mediated neurotoxicity remain poorly understood. Using whole-cell patch clamp recording in cultured hippocampal neurons, this study found that gp120 significantly increased the outward delayed-rectifier K+ currents (Ik). Meanwhile, Western blot assay revealed that gp120 markedly upregulated Kv2.1 protein levels, which was consistent with the increased Ik density. With Western blot and terminal deoxynucleotidyl transferase dUTP nick end labeling assays, it was discovered that gp120-induced neuronal injury was largely due to activation of Kv2.1 channels and resultant apoptosis mediated by caspase-3 activation, as the pharmacological blockade of Kv2.1 channels largely attenuated gp120-induced cell damage and caspase-3 expression. Moreover, p38 MAPK was demonstrated to participate in gp120-induced hippocampal neural damage, since p38 MAPK antagonist (SB203580) partially abrogated gp120-induced Kv2.1 upregulation and neural apoptosis. Taken together, these results suggest that gp120 induces hippocampal neuron apoptosis by enhancement of the Ik, which might be associated with increased Kv2.1 expression via the p38 MAPK pathway.”
Protein phosphorylation is the major molecular mechanism through which protein function is regulated in response to extracellular stimuli both inside and outside the nervous system. Virtually all types of extracellular signals, including neurotransmitters, hormones, light, neurotrophic factors and cytokines, produce most of their diverse physiological effects by regulating phosphorylation of specific phosphoproteins in their target cells. Virtually every class of neuronal protein is regulated by phosphorylation.
Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1 (2007)
Abstract
Kv2.1, the primary delayed rectifying potassium channel in neurons, is extensively regulated by phosphorylation. Previous reports have described Kv2.1 phosphorylation events affecting channel gating and the impact of this process on cellular excitability. Kv2.1, however, also provides the critical exit route for potassium ions during neuronal apoptosis via p38 MAPK-dependent membrane insertion, resulting in a pronounced enhancement of K+ currents.
Here, electrophysiological and viability studies using Kv2.1 channel mutants identify a p38 phosphorylation site at Ser-800 (S800) that is required for Kv2.1 membrane insertion, K+ current surge, and cell death. In addition, a phospho-specific antibody for S800 detects a p38-dependent increase in Kv2.1 phosphorylation in apoptotic neurons and reveals phosphorylation of S800 in immunopurified channels incubated with active p38. Consequently, phosphorylation of Kv2.1 residue S800 by p38 leads to trafficking and membrane insertion during apoptosis, and remarkably, the absence of S800 phosphorylation is sufficient to prevent completion of the cell death program.
But wait, we aren't quite finished yet. From the previously discussed paper we know that p38 MAPK participates in gp120-induced hippocampal neural damage.
What are the signalling pathways inducing this?
Surprise surprise, it's a now familiar pathway. But first some background on MAPK. “Complex” doesn't begin to describe it:
What is MAPK Signaling Pathway?
Mitogen-activated protein kinase (MAPK) modules containing three sequentially activated protein kinases are key components of a series of vital signal transduction pathways that regulate processes such as cell proliferation, cell differentiation, and cell death in eukaryotes from yeast to humans.
(KY-nays) A type of enzyme (a protein that speeds up chemical reactions in the body) that adds chemicals called phosphates to other molecules, such as sugars or proteins. This may cause other molecules in the cell to become either active or inactive. Kinases are a part of many cell processes.
The mitogen-activated protein kinase (MAPK) cascade is a highly conserved module that is involved in various cellular functions, including cell proliferation, differentiation and migration. The pathway covers many proteins, including MAPK originally named ERK (extracellular signal-regulated kinases), which transmits signal by phosphorylates a neighboring protein, as like an "on" or "off" switch.
The Function of MAPK Signaling Pathway
The MAPK signaling pathway is essential in regulating many cellular processes including inflammation, cell stress response, cell differentiation, cell division, cell proliferation, metabolism, motility and apoptosis. The role of the MAPK pathway in cancer, immune disorders and neurodegenerative diseases has been well recognized.
As an aside I got a chat box popup on their website and thanked the nice lady for posting the schematic and letting me use it.
An excellent paper from 2021, full of red flags and worth reading in full:
SARS-CoV-2 Spike Protein Elicits Cell Signaling in Human Host Cells: Implications for Possible Consequences of COVID-19 Vaccines
Abstract
The world is suffering from the coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 uses its spike protein to enter the host cells. Vaccines that introduce the spike protein into our body to elicit virus-neutralizing antibodies are currently being developed. In this article, we note that human host cells sensitively respond to the spike protein to elicit cell signaling. Thus, it is important to be aware that the spike protein produced by the new COVID-19 vaccines may also affect the host cells. We should monitor the long-term consequences of these vaccines carefully, especially when they are administered to otherwise healthy individuals. Further investigations on the effects of the SARS-CoV-2 spike protein on human cells and appropriate experimental animal models are warranted.
On p38 activation, which plays a key part in neurotoxicity:
The SARS-CoV-2 spike protein without the rest of the viral components has also been shown to activate cell signaling by Patra et al. [29]. The authors reported that the full-length SARS-CoV-2 spike protein expressed by the means of transient transfection, either in the human lung alveolar epithelial cell line A549 or in the human liver epithelial cell line Huh7.5, activated NF-κB and AP-1 transcription factors as well as p38 and ERK mitogen-activated protein kinases, releasing interleukin-6. This cell signaling cascade was found to be triggered by the SARS-CoV-2 spike protein downregulating the ACE2 protein expression, subsequently activating the angiotensin II type 1 receptor [29]. These experiments using transient transfection may reflect the intracellular effects of the spike protein that could be triggered by the RNA- and viral vector-based vaccines.
These results collectively reinforce the idea that human cells are sensitively affected by the extracellular and/or intracellular spike proteins though the activation of cell signal transduction.
Recall that once cleaved, serum spike S1 can cross the blood brain barrier, enter an exosome and be endocytosed without needing to bind to a receptor like ACE2.
DC-SIGN (Dendritic Cell-specific Intercellular adhesion molecule-3-Grabbing Non-integrin) is a C-type lectin receptor present on the surface of both macrophages and dendritic cells which binds to GP120.
In the context of mutations being driven in cleaved S1 Vs in the more genomically stable S2:
More critically to brain pathology, it also binds totubilin beta III, a component of neuronal microtubules and reducesacetylation of α-tubulin.
…gp120 impairs tubulin function by binding to tubulin isoforms and inhibiting proper assembly of microtubules. In this manner, gp120 can then disrupt normal axonal transport that is critical for neuronal maintenance and survival.
Note that changes to the neuronal cytoskeletal structure and function persist after the inflammation has gone. Is this a hypothesis to explain persistent brain fog in some individuals suffering from “long Covid”?
Are Microtubules the Brain of the Neuron (2015)
The control of the microtubule function is extremely complex, establishing and maintaining the architecture of the neuron. The control is distributed throughout the cell, which brings up the question of where the central control is to respond to mental events in such detailed and complex ways. They are highly regulated in terms of the number, length, allocation, exact positioning and placement. Abnormalities in any of these functions leads to brain disease. Mutations in the proteins that hold the microtubules in place, such as tau are critical for the development of major neurodegenerative diseases (see post). The motors that move material along the microtubule highways—dyneins and kinesins—and regulators of these motors—dynactins—can produce other brain diseases. Defective transport in the axon is associated with ALS, Alzheimer’s, and multiple sclerosis.
A kinesin motor protein dragging a vesicle along a microtubule:
Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production (2014)
In this report, we present evidence that microtubule acetylation is a critical component of innate immunity. Microtubule acetylation is induced by bacterial lipopolysaccharides (LPS) treatment and selectively required for the production of the anti-inflammatory cytokine IL-10. Modulation of tubulin acetylation by targeting MEC17 and HDAC6 profoundly affects IL-10 production and anti-inflammatory activity in macrophages and in mice. We further show that microtubule acetylation selectively enhances p38 signalling, leading to SP-1 dependent IL-10 transcription.
The viral protein gp120 decreases the acetylation of neuronal tubulin: potential mechanism of neurotoxicity (2017)
Abstract
The human immunodeficiency virus (HIV) envelope protein gp120 promotes axonal damage and neurite pruning, similar to that observed in HIV-positive subjects with neurocognitive disorders. Thus, gp120 has been used to examine molecular and cellular pathways underlying HIV-mediated neuronal dysfunction. Gp120 binds to tubulin beta III, a component of neuronal microtubules. Microtubule function, which modulates the homeostasis of neurons, is regulated by polymerization and post-translational modifications. Based on these considerations, we tested the hypothesis that gp120 induces dynamic instability of neuronal microtubules. We first observed that gp120 prevents the normal polymerization of tubulin in vitro. We then tested whether gp120 alters the post-translational modifications in tubulin by examining the ability of gp120 to change the levels of acetylated tubulin in primary rat neuronal cultures. Gp120 elicited a time-dependent decrease in tubulin acetylation that was reversed by Helix-A peptide, a compound that competitively displaces the binding of gp120 to neuronal microtubules. To determine whether post-translational modifications in tubulin also occur in vivo, we measured acetylated tubulin in the cerebral cortex of HIV transgenic rats (HIV-tg). We observed a decrease in tubulin acetylation in 5- and 9-month-old HIV-tg rats when compared to age-matched wild type. Neither changes in microglia morphology nor alterations in mRNA levels for interleukin-1β and tumor necrosis factor α were detected in 5-month-old animals. Our findings propose neuronal microtubule instability as a novel mechanism of HIV neurotoxicity, without evidence of enhanced inflammation.
Keywords: HAND; HIV transgenic rats; Helix-A peptide; microglia; microtubules.
In considering the potential interaction between gp120 and MTs as an underlying contributing factor to neuronal dysfunction in HAND, we examined the ability of gp120 to interfere with MT polymerization and change tubulin acetylation. We report that gp120 inhibits normal MTs polymerization and alters typical PTM patterning. We propose that synaptic injury seen in HAND could be caused by gp120 ability to negatively affect neuronal cytoskeletal structure and function.
Discussion
Despite the lack of direct infection, neurons remain a prime target for the toxic adverse effects of HIV. Knowledge of the mechanisms whereby HIV causes neurodegeneration is invaluable in understanding HAND-associated neuropathology. Although massive neuronal loss may be responsible for the dementia observed in severe cases of HAND, it appears that neuronal apoptosis is a late event and does not represent the main pathological substrate of asymptomatic or mild HAND (Clifford and Ances 2013). Furthermore, the relatively rapid onset and progression of neurological decline does not support neuronal loss as the primary cause of HAND. Therefore, HAND more likely reflects a specific neuronal dysfunction resulting from the combined effects of several mechanisms. Our data suggest that HIV, through gp120, causes an initial “traumatic” event in axons and dendrites that may culminate in retrograde degeneration and synaptic simplifications seen in HAND.
It is well established that gp120 is internalized by neurons (Bachis et al. 2006; Berth et al. 2015), binds to mannose-binding lectin (Teodorof et al. 2014), and is retrogradely transported along the axon to the soma (Bachis et al. 2006). During axonal transport, gp120 binds with high affinity to the carboxy terminal of at least two neuronal-specific tubulin isoforms, TUBB3 and Δ2 isoform of TUB α1/β1 (Avdoshina et al. 2016b). Here, we show that gp120 reduces MT polymerization and acetylation of α-tubulin, suggesting that gp120 binding to MTs results in the destabilization of the cytoskeleton and its function. These results are corroborated by the finding that gp120 decreases the ability of MTs to transport mitochondria (Avdoshina et al. 2016a) in neurons. Abnormal mitochondrial transport has been implicated in the progression of multiple neurodegenerative diseases, including Huntington’s (Chang et al. 2006), Parkinson’s (Wild and Dikic 2010), and Alzheimer’s disease (Anandatheerthavarada et al. 2003). However, altered mitochondria transport in these diseases is brought about by different molecular and cellular mechanisms. For instance, in Huntington’s disease, mutated huntingtin inhibits axonal transport by acting at kinesin-1 motor protein (Morfini et al. 2009) or by interacting with mitochondrial fission GTPase, which regulates mitochondrial transport (Song et al. 2011). Our data suggest that in HAND, gp120 impairs tubulin function by binding to tubulin isoforms and inhibiting proper assembly of MTs. In this manner, gp120 can then disrupt normal axonal transport that is critical for neuronal maintenance and survival.
In this study, we provide evidence that the HIV envelope protein interferes with normal regulation of the cytoskeleton, both by inhibiting MT polymerization and reducing acetylation of tubulin in neurons. These changes may reduce MT functionality, including trafficking of mitochondria (Avdoshina et al. 2016a), which is crucial for neuronal survival (Belzil et al. 2013; Franker and Hoogenraad 2013; Nahm et al. 2013). We also show that lower levels of Ac-tubulin are present in the cortex of HIV-tg rats when compared to wt even in the absence of a concurrent neuroinflammatory response as measured by cytokine production and changes in microglia morphology. Thus, it appears that the alteration in cytoskeleton function observed in our studies precedes the ongoing neuroinflammation that has been previously proposed as one of the mechanisms of HAND. This consideration may explain why HAND still persists even though the antiretroviral therapy has reduced the cases of encephalitis (Gelman 2015). Thus, we propose that the direct interaction of gp120 with components of the cytoskeleton could significantly contribute to the loss of synaptic connectivity and the slow but progressive neurodegeneration seen in HAND subjects.
Helix-A peptide, which has been shown to displace gp120 from binding to TUBB3, prevents the degeneration of neuronal processes as well as improves neuronal survival (Avdoshina et al. 2016b). In this study, we show that Helix-A peptide reduces the ability of gp120 to decrease Ac-tubulin. Our data provide additional evidence that gp120 binding to TUBB3 promotes neuronal degeneration by disruption of MT stability. The role of decreased Ac-tubulin in the overall neurotoxic effect of gp120, however, is, at present, only speculative. Although acetylation of tubulin is frequently associated with stable MTs, acetylation itself does not stabilize MTs (Haggarty et al. 2003). Nevertheless, acetylation at Lys 40 of α-tubulin plays a role in recruiting molecular motors such as kinesin-1 and dynein/dynactin (Reed et al. 2006; Dompierre et al. 2007), suggesting that acetylation of tubulin activates both anterograde and retrograde trafficking. Whether gp120 inhibits kinesin-1 or other molecular motors still remains a question. In addition, tubulin acetylation may decrease binding of tubulin-associated proteins, such as Tau, which affects microtubule polymerization in neurons. (Popov et al. 2001; Fauquant et al. 2011; Janning et al. 2014). Clearly, this is a complex mechanism that warrants further investigation.
Tunneling nanotubes provide a route for SARS-CoV-2 spreading
A paper published 20th July ‘22 using advanced cryo-correlative light and electron microscopy (CLEM) and cryo-electron tomography (ET) gives further weight to this hypothesis. The virus or spike protein is neurotoxic not just by disrupting microtubules but by also mediating cell to cell connections:
The virus or spike protein is able to enter the neuron without binding to ACE2 receptors.
Viral protein fragments such as spike being trafficked between cells along tunneling nanotubes (TNT), either inside or on the surface.
Motor proteins being proposed as a ferrying mechanism.
p38-MK2 mediated TNT formation.
These are short lived, 30-60 seconds before depolymerising, which is long enough to allow transportation to the neighbouring cell.
Scientists identify key role of p38-MK2 pathway in determining cell fate in response to stress
…When cells are exposed to high levels of stress, the p38-MK2 pathway is activated in a sustained manner, promoting the degradation of the MK2 protein, which correlates with cell death. However, moderate levels of stress trigger only a temporary activation of the p38-MK2 pathway, which allows the re-accumulation of MK2 and leads to cell survival. Thus, MK2 protein levels act as a molecular indicator that informs cells whether to stay alive or to initiate self-destruction.
Tunneling nanotubes provide a route for SARS-CoV-2 spreading
Abstract
Neurological manifestations of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection represent a major issue in long coronavirus disease. How SARS-CoV-2 gains access to the brain and how infection leads to neurological symptoms are not clear because the principal means of viral entry by endocytosis, the angiotensin-converting enzyme 2 receptor, are barely detectable in the brain. We report that human neuronal cells, nonpermissive to infection through the endocytic pathway, can be infected when cocultured with permissive infected epithelial cells. SARS-CoV-2 induces the formation of tunneling nanotubes (TNTs) and exploits this route to spread to uninfected cells. In cellulo correlative fluorescence and cryo–electron tomography reveal that SARS-CoV-2 is associated with TNTs between permissive cells. Furthermore, multiple vesicular structures such as double-membrane vesicles, sites of viral replication, are observed inside TNTs between permissive and nonpermissive cells. Our data highlight a previously unknown mechanism of SARS-CoV-2 spreading, likely used as a route to invade nonpermissive cells and potentiate infection in permissive cells.
This evidence supports our hypothesis that SARS-CoV-2, similar to other viruses such as HIV (30, 72), is an inducer of TNT formation, to facilitate its spreading between TNT-connected cells. SARS-CoV-2 might be able to induce TNT formation via several mechanisms.
(C) Coculture was stained with the anti-N and anti-S antibodies to detect the virus. The gray arrows indicate the SARS-CoV-2 anti-N signal, and the green arrows indicate the anti-S signal inside TNT.
The yellow arrow points the TNT. (D and E) 2D confocal micrograph (D) and 3D rendering (E) showing a TNT connecting two SH-SY5Y mCherry cells, cocultured with infected Vero E6 cells. The yellow arrow points the TNT between the SH-SY5Y mCherry cells. The white arrows indicate SARS-CoV-2 inside the TNT. Cellular cytoplasm and TNTs were labeled with CellMask Blue. Scale bars, 10 μm
DISCUSSION
Patients with COVID-19 exhibit a range of neurological symptoms, suggesting that SARS-CoV-2 can invade the CNS (64). After an autopsy of the brains of patients with COVID-19, CoV RNA has been detected (65), and the olfactory mucosa has been suggested as a route of viral entry (19). SARS-CoV-2 is known to infect human host cells by binding ACE2, of which the expression is low in neuronal cells (27). Nonetheless, several studies have reported the presence of SARS-CoV-2 in human pluripotent stem cell and primary neurons (5, 66). Thus, how SARS-CoV-2 could enter neuronal cells is still an open question.Several viruses, such as the influenza virus, HIV, and herpes simplex virus (31), can use TNTs to transfer their genomes to naïve cells, a mechanism of direct cell-to-cell communication that allows evasion of host immunity and to avoid pharmaceutical targeting (67). Here, we demonstrate that human neuronal SH-SY5Y cells, nonpermissive to SARS-CoV-2 through an exocytosis/endocytosis-dependent pathway (fig. S1), can be infected after being cocultured with permissive Vero E6 epithelial cells previously infected with SARS-CoV-2 (Fig. 1). Furthermore, our data using remdesivir and immunostaining for the viral replicative markers J2 and nsp3 support that SARS-CoV-2 is able to replicate once inside neuronal cells (Fig. 2 and fig. S5). By blocking the ACE2-mediated entry of the virus with a neutralizing antibody, we also demonstrate that SARS-CoV-2 can spread between permissive cells through a secretion-independent pathway.We speculate that TNTs accelerate the propagation of the infection, even between permissive cells. Because TNTs are dynamic transient structures (28) where actin is able to polymerize and depolymerize rapidly (i.e., 30 to 60 s) (68, 69), the virus could potentially spread faster through TNTs than through other routes (67). Previous evidence indicated that upon interaction with cellular protrusions, viruses undergo rapid actin- and myosin II–mediated transport by “surfing” on the cell surface before reaching entry sites closer to the cell body (60). Moreover, TNTs may contain unconventional actin-based myosin motor proteins such as myosin Va (MyoVa) and MyoX (28, 70). MyoVa has been suggested to mediate an actomyosin-dependent transport of endocytic vesicles in TNTs (28), while MyoX has been proposed as a major player in TNT formation (71).
Notably, SARS-CoV-2 infection resulted in an increase in the percentage of TNT-connected cells both between Vero E6 cells and between Vero E6 and SH-SY5Y cells (Fig. 5 and fig. S7). This evidence supports our hypothesis that SARS-CoV-2, similar to other viruses such as HIV (30, 72), is an inducer of TNT formation, to facilitate its spreading between TNT-connected cells. SARS-CoV-2 might be able to induce TNT formation via several mechanisms. A recent publication has shown that SARS-CoV-2 infection induces a marked increase in filopodial protrusions, a process in which casein kinase II (CK2) plays a role (63). CK2 activity was significantly up-regulated in SARS-CoV-2–infected Vero E6 cells (63). CK2 might also be involved in the increase in TNTs, as it promotes actin polymerization and regulates the organization of the cytoskeleton (73). CK2 is known to phosphorylate myosin proteins at endocytic sites to drive actin polymerization (74). For example, Marburg virus hijacks the unconventional motor protein MyoX, which promotes filopodia formation and the traffic of the virus along them (31). We have previously shown that MyoX is a positive regulator of TNT formation in neuronal cells (71). It would be interesting to investigate whether MyoX is also involved in the TNT formation induced by SARS-CoV-2 and its movement along TNTs.
In addition, Bouhaddou et al. (63) showed the activation of the p38 mitogen-activated protein kinase (MAPK) signaling pathway after SARS-CoV-2 cell infection. Activation of the p38 MAPK could also increase TNT formation (75). By confocal microscopy, we detected the viral proteins (S and N) and the replicative marker (J2 and nsp3) within TNTs, suggesting that TNTs could transfer viral materials. To better investigate how SARS-CoV-2 transfer through TNTs, we set up a challenging approach called CLEM, cryo-EM, and cryo-ET (39). These techniques allowed us to assess (in correlative mode) both SARS-CoV-2 and TNT architecture under the closest to native conditions. We found multiple SARS-CoV-2 virions (detected using an anti-S antibody) associated with the PM of TNTs formed between permissive cells (Figs. 6 and 7). We also observed the vesicular structures of different sizes in correspondence with the fluorescent signal of the virions inside TNTs. Similar viral vesicular structures and DMVs were present inside TNTs between permissive and nonpermissive cells (Fig. 8). Notably, we observed the virus on top of the TNTs formed between permissive cells and not in heterotypic coculture with neuronal cells. This discrepancy could be explained by the presence of the ACE2 receptor only on the TNT membranes derived from Vero E6 cells and not from SH-SY5Y cells. However, the lipid composition of the TNT membranes might also vary between different cell types. Because we observed that SARS-CoV-2 particles adhere to the surface of cell protrusions (i.e., TNTs) connecting two permissive cells, we hypothesize that SARS-CoV-2 might “surf” on the cell membrane. On the basis of our observations, the transfer of SARS-CoV-2 occurs via TNTs through both extracellular adhesion (i.e., surfing) and intracellular transport in agreement with what has been already shown for HIV (30).
In conclusion, here, we show that SARS-CoV-2 is able to hijack TNTs to spread between connected cells, indicating that this intercellular route could contribute to the pathogenesis of COVID-19 and the spreading of the virus to nonpermissive neuronal cells. Within the limitation of the cellular model used, our report provides unprecedented structural information of SARS-CoV-2 by cryo-CLEM and cryo-ET and how it might use TNTs for spreading between permissive and nonpermissive cells to increase both viral tropism and infection efficiency. These results also pave the way to further investigations of the role of cell-to-cell communication in SARS-CoV-2 spreading to the brain in more physiological contexts (e.g., the potential role of TNTs in the spreading of the virus from the olfactory epithelium of the nasal cavity to the olfactory sensory neurons in the CNSand in contributing to the occurrence of the long COVID syndrome) and on alternative therapeutic approaches to impairing viral diffusion in addition to current investigations mainly focused on blocking S-receptor interactions.
There is more, as touched on previously - a parallel MAPK inflammatory pathway mediated by interleukin 6 (IL-6), also promoted by spike protein and lipid nanoparticles.
That's a lot of potential pathogenic inflammation in the brain.
SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells (2020)
SARS-CoV-2 spike protein mediated activation of AT1 signaling induces MAPK/NF-κB axis
We examined the possibility that AT1 upregulation may play a role in MAPK and NF-kB activation in virus infected or spike gene transfected cells generated from two distinct human tissues (Huh7.5 and A549). We observed that SARS-CoV-2 infected Huh7.5 and A549 cells expressed an elevated level of phospho-p38 MAPK (Thr180/Tyr182) and phospho-p42/44 MAPK (Thr202/Tyr204) molecules (Fig 2A and 2B). MAPK regulates p65/NF-κB activation for cytokine synthesis.
Fig 2. SARS-CoV-2 spike protein activates the transcription factors for IL-6 synthesis.
(A, B) Western blot analysis of phospho-p38 MAPK (Thr180/Tyr182) and phospho-p42/44 MAPK (Thr202/Tyr204) in Huh7.5 and A649 cell lysates prepared after 48 h of mock- or SARS-CoV-2 virus infection, or transiently transfected with empty vector or SARS-CoV-2 spike gene construct. (C, D) Western blot analysis of phospho-NF-κB (Ser276), IκBα and c-Fos expression in Huh7.5 and A549 cell lysates prepared after 48 h of mock- or SARS-CoV-2 virus infection, or transiently transfected with empty vector or SARS-CoV-2 spike gene construct. Expression level of actin or tubulin in each lane from the same gel is shown as a total protein load for comparison.
Fig 3. Candesartan cilexetil as an AT1 receptor antagonist prevent MAPK activation.
(A) Western blot analysis of phospho-p38 MAPK (Thr180/Tyr182) and phospho-p42/44 MAPK (Thr202/Tyr204) in Huh7.5 cell lysates prepared after 48 h of transfection of SARS-CoV-2 spike gene construct with or without Candesartan cilexetil treatment. Expression level of actin in each lane is shown as a total protein loading control for comparison. (B) Western blot analysis of phospho-p38 MAPK (Thr180/Tyr182) and phospho-p42/44 MAPK (Thr202/Tyr204) in A549 cell lysates prepared after 48 h of transfection of SARS-CoV-2 spike gene construct with or without Candesartan cilexetil treatment. Expression level of actin or tubulin in each lane from the same gel is shown as a total protein load for comparison.
So what diseases may be caused by neurotoxic Kv2.1 protein, now we know that gp120 markedly upregulates expression?
If you want the blue pill, leave now and go watch Netflix or something.
For the red pill it's over to Wiki…
KCNB1
Modulation of K+ channel activity and expression has been found to be at the crux of many profound pathophysiological disorders in several cell types.
Neurodegenerative Disease
Oxidative damage is widely considered to play a role in neurodegenerative disorders, including Alzheimer's disease. Such oxidative stress alters the redox sensitivity of the Kv2.1 delayed rectifier, resulting in the modulation of the channel.[8] In vitro studies and studies in animal models show that when KCNB1 is oxidized, it no longer conducts, leading to neurons becoming hyperpolarized and dying; oxidized KCNB1 also clusters in lipid rafts and cannot be internalized, which also leads to apoptosis. These alterations disrupt normal neuronal signaling and increase the likelihood of neurological diseases. Oxidized (oligomerized) KCNB1 channels are present in the hippocampi of old (Braak stage 1-2) and Alzheimer's disease (Braak stage 5) donors of either sexes [20][23]
As indicated earlier, oxidative and nitrosative injurious stimuli also activate a cell death-inducing cascade that promotes to a zinc and calcium/clamodulin-dependent interaction between syntaxin and Kv2.1, leading to the pro-apoptotic insertion of additional potassium channels into the plasma membrane. These new population of channels aid in the loss of intracellular potassium, creating a permissive environment for protease and nuclease activation in injured neurons.[8] Agents that interfere with the Kv2.1/syntaxin interaction are highly neuroprotective in acute ischemic injury models (stroke) [24]
Increased probability of the channel remaining open can also potentially drive neurodegeneration. Human immunodeficiency virus type-1 (HIV-1)-associated dementia (HAD) may be driven by an overabundance of glutamate, which in turn can trigger increased calcium levels, which in turn can drive calcium-dependent dephosphorylation of KCNB1 channels, which increases probability of channel activation and current conductance. Enhanced KCNB1 current couples cell shrinkage associated with apoptosis and dendritic beading leading to diminished long term potentiation. These neuronal modifications may explain the atrophy of cell layer volume and late stage cell death observed in HAD disease.[25]
Cancer
Exploitation of this channel is advantageous in cancer cell survival as they have the ability to produce heme oxygenase-1, an enzyme with the ability to generate carbon monoxide (CO). Oncogenic cells benefit from producing CO due to the antagonizing effects of the KCNB1 channel. Inhibition of KCNB1 allows cancer proliferation without the apoptotic pathway preventing tumor formation. Although potassium channels are studied as a therapeutic target for cancer, this apoptotic regulation is dependent on cancer type, potassium channel type, expression levels, intracellular localization as well as regulation by pro- or anti-apoptotic factors.[26]
Another name for Human immunodeficiency virus type-1 (HIV-1)-associated dementia (HAD)isAIDS Dementia:
HIV and AIDS Dementia (2021)
Decline in mental processes is a complication of advanced HIV infection.
Although the specific symptoms vary from person to person, they may be part of a single disorder known as AIDS dementia complex, or ADC. Other names for ADC are HIV-associated dementia and HIV/AIDS encephalopathy.
Common symptoms include decline in thinking, or "cognitive," functions such as memory, reasoning, judgment, concentration, and problem solving.
Other common symptoms are changes in personality and behavior, speech problems, and motor (movement) problems such as clumsiness and poor balance.
When these symptoms are severe enough to interfere with everyday activities, a diagnosis of dementia may be warranted.
AIDS dementia complex typically occurs as the CD4+ count falls to less than 200 cells/microliter. It may be the first sign of AIDS. With the advent of antiretroviral therapy (ART), the frequency of ADC has declined. ART may not only prevent or delay the onset of AIDS dementia complex in people with HIV infection, it can also improve mental function in people who already have ADC.
Causes of AIDS Dementia
AIDS dementia complex is caused by the HIV virus itself, not by the opportunistic infections that occur commonly in advanced HIV. We do not know exactly how the virus damages brain cells.
HIV may affect the brain through several mechanisms. Viral proteins may damage nerve cells directly or by infecting inflammatory cells in the brain and spinal cord. HIV may then induce these cells to damage and disable nerve cells. HIV causes generalized inflammation, which can cause memory issues, as well ass other aging processes, including heart disease.
Symptoms of AIDS Dementia
AIDS dementia complex can affect behavior, memory, thinking, and movement. At first, symptoms are subtle and may be overlooked, but they gradually become troublesome. The symptoms vary widely from person to person.
Symptoms of early dementia include:
Reduced productivity at work
Poor concentration
Mental slowness
Difficulty learning new things
Changes in behavior
Decreased libido
Forgetfulness
Confusion
Word-finding difficulty
Apathy (indifference)
Withdrawal from hobbies or social activities
Depression
Initially subtle disease can progress to more severe symptoms such as:
Sleep disturbances
Psychosis -- Severe mental and behavioral disorder, with features such as extreme agitation, loss of contact with reality, inability to respond appropriately to the environment, hallucinations, delusions
Mania -- Extreme restlessness, hyperactivity, very rapid speech, poor judgment
Seizures
Without ART, these symptoms gradually worsen. They can lead to a vegetative state, in which the person has minimal awareness of his or her surroundings and is incapable of interacting.
Media file 1: CT scan of the brain of a patient with AIDS dementia complex (ADC) shows diffuse atrophy (loss of tissue) and ventricular enlargement and attenuation (dark areas) around the ventricles in the white matter.
Media file 4: Photomicrograph from patient with AIDS dementia complex (ADC) illustrates the intense astrogliosis (scarring) that is characteristic of HIV encephalitis. Contributed by Dr. Beth Levy, Saint Louis University School of Medicine, St. Louis.
In 2016, Berth et al used rat dorsal root ganglions (DRG) and isolated squid axoplasm to investigate the intra-axonal effects of gp120 on fast axonal transport (FAT). Pathology resulting from this for HIV patients can include sensory neuropathies, including distal sensory polyneuropathy (DSP).
Gp120 doesn’t necessarily cause the immediate loss of the neuron via apoptosis or immune signalling responses but instead may impair its functioning long term.
Sensory neuropathies refer to a host of diseases that result in loss of sensation throughout the body. Collectively, sensory neuropathies can result from a plethora of conditions that this review will discuss. These may further sub-divide into small fiber (pain-dominant) and large fiber (ataxia-predominant) pathologies.
Symptoms of distal sensory polyneuropathy (DSP) include tingling, numbness, or burning pain that normally begins in the legs and feet and may spread to the hands. There are two types of DSP: HIV-DSP, which is due to HIV infection itself and antiretroviral (ARV)-DSP, which is caused by certain ARV drugs.
An axon (from Greek ἄξων áxōn, axis), or nerve fiber (or nerve fibre: see spelling differences), is a long, slender projection of a nerve cell, or neuron, in vertebrates, that typically conducts electrical impulses known as action potentials away from the nerve cell body. The function of the axon is to transmit information to different neurons, muscles, and glands. In certain sensory neurons (pseudounipolar neurons), such as those for touch and warmth, the axons are called afferent nerve fibers and the electrical impulse travels along these from the periphery to the cell body and from the cell body to the spinal cord along another branch of the same axon. Axon dysfunction can be the cause of many inherited and acquired neurological disorders that affect both the peripheral and central neurons. Nerve fibers are classed into three types – group A nerve fibers, group B nerve fibers, and group C nerve fibers. Groups A and B are myelinated, and group C are unmyelinated. These groups include both sensory fibers and motor fibers. Another classification groups only the sensory fibers as Type I, Type II, Type III, and Type IV.
Axonal transport, also called axoplasmic transport or axoplasmic flow, is a cellular process responsible for movement of mitochondria, lipids, synaptic vesicles, proteins, and other organelles to and from a neuron's cell body, through the cytoplasm of its axon called the axoplasm. Since some axons are on the order of meters long, neurons cannot rely on diffusion to carry products of the nucleus and organelles to the end of their axons. Axonal transport is also responsible for moving molecules destined for degradation from the axon back to the cell body, where they are broken down by lysosomes.
Dynein, a motor protein responsible for retrograde axonal transport, carries vesicles and other cellular products toward the cell bodies of neurons. Its light chains bind the cargo, and its globular head regions bind the microtubule, "inching" along it.
Movement toward the cell body is called retrograde transport and movement toward the synapse is called anterograde transport.
Sensory neuropathies are the most common neurological complication of HIV. Of these, distal sensory polyneuropathy (DSP) is directly caused by HIV infection and characterized by length-dependent axonal degeneration of dorsal root ganglion (DRG) neurons. Mechanisms for axonal degeneration in DSP remain unclear, but recent experiments revealed that the HIV glycoprotein gp120 is internalized and localized within axons of DRG neurons. Based on these findings, we investigated whether intra-axonal gp120 might impair fast axonal transport (FAT), a cellular process critical for appropriate maintenance of the axonal compartment. Significantly, we found that gp120 severely impaired both anterograde and retrograde FAT. Providing a mechanistic basis for these effects, pharmacological experiments revealed an involvement of various phosphotransferases in this toxic effect, including members of mitogen-activated protein kinase pathways (Tak-1, p38, and c-Jun N-terminal Kinase (JNK)), inhibitor of kappa-B-kinase 2 (IKK2), and PP1. Biochemical experiments and axonal outgrowth assays in cell lines and primary cultures extended these findings. Impairments in neurite outgrowth in DRG neurons by gp120 were rescued using a Tak-1 inhibitor, implicating a Tak-1 mitogen-activated protein kinase pathway in gp120 neurotoxicity. Taken together, these observations indicate that kinase-based impairments in FAT represent a novel mechanism underlying gp120 neurotoxicity consistent with the dying-back degeneration seen in DSP. Targeting gp120-based impairments in FAT with specific kinase inhibitors might provide a novel therapeutic strategy to prevent axonal degeneration in DSP.
Figure 6. Gp120 impairs neurite outgrowth through a Tak1-dependent mechanism.
Primary rat dorsal root ganglion (DRG) neurons were treated with gp120 to determine whether gp120 altered neuronal morphology via the ability to grow or maintain neurite extensions. Representative photomicrographs illustrate DRG neurons that were untreated (a), treated with 2 nM gp120 (b), 2 nM gp120 plus 15 nM (5 Z)-7-oxozeaenol (c), or 2 nM heat-inactivated gp120 (d). Scale bars indicate 100 µm. The average percentage of DRG neurons exhibiting neurite extensions (e); calculated from separating the DRG neurons into two categories, one included counts of DRG neurons that displayed neurites and the other included counts of DRG neurons that lacked neurites. The average neurite outgrowth length per DRG neuron was determined by measuring all neurites within the sample and dividing by the number of DRG neurons that displayed neurites (f), results given in micrometers. All results averaged from three independent experiments that examined 3,154 neurons in total, including 2,374 neurons exhibiting neurites and 780 neuronal cell bodies without neurites. *p < .05 compared with untreated control, gp120 + (5 Z)-7-oxozeaenol, and heat-inactivated gp120 groups.
Figure 7. Diagram depicting kinase pathways activated by gp120.
The use of pharmacological inhibitors in axonal transport studies here delineated novel signaling pathways by which gp120 impairs FAT. Gp120 was found to activate, directly or indirectly (dotted line) the MAP3K Tak1. Tak1 in turn activates at least two independent signaling pathways. In one pathway, Tak1 activates the IKK complex through a mechanism that requires PP1 activity and leads to activation of IKK-2. How IKK-2 inhibits FAT is currently unknown and depicted by the dotted line. In the second pathway, Tak1 activates canonical MAP2Ks which in turn, promote activation of both p38 MAPK and JNK. Both p38 MAPK and JNK are known to phosphorylate kinesin to inhibit FAT. (5 Z)-7-Oxozeaenol: Tak-1 inhibitor. DVD peptide blocks conserved docking domains to inhibit certain MAP3K's. Okadaic acid inhibits serine or threonine phosphatases. I-2: specific inhibitor of the serine or threonine phosphatase PP1. Inhibitor XII: a specific inhibitor of IKK-2. MW069A: a specific inhibitor of p38 MAPK. SP600125: a specific inhibitor of JNK. SB203580: an inhibitor of p38 MAPK and JNK3.
Discussion
Although DSP is the most prevalent neurological complication of HIV, affecting close to 30% of patients, its pathogenesis remains unclear and treatments are limited (Phillips et al., 2010). Proposed pathological mechanisms of DSP include indirect neurotoxicity by activation of macrophages and direct neurotoxicity by release of viral proteins, such as gp120, which interact with DRG neurons. A recent study indicated that gp120 is internalized by DRG neurons and localizes within their axons (Berth et al., 2015), suggesting the possibility of an intra-neuronal mechanism for gp120 to cause neurotoxicity.
How might intra-neuronal gp120 cause the axonal degeneration phenotype seen in DSP? One possible mechanism is through impairments in FAT. Loss-of-function mutations of the motor proteins kinesin or CDyn are sufficient to cause dying-back neuropathies (Reid et al., 2002; Puls et al., 2003; Munch et al., 2005; Ebbing et al., 2008; Dupuis et al., 2009). Since the concentration of kinesin is 0.5 µM and tubulin is 25 µM in squid axoplasm (Morris and Lasek, 1984; Brady et al., 1990), the concentration of perfused gp120 was not high enough to cause sequestration of kinesin or impair FAT through binding to microtubules (Brady et al., 1990). Further, gp120 perfusion did not alter microtubule structure in the squid axoplasm (data not shown). Therefore, a more plausible explanation is that the severe effect on FAT elicited by gp120 results from activation of signaling cascades that regulate FAT.
Since the amount of free gp120 in plasma has been determined to be in the low picomolar to low nanomolar range (Gilbert et al., 1991; Oh et al., 1992), 100 pM gp120 was perfused to determine if a lower, more physiological relevant concentration might affect transport. In fact, 100 pM gp120 impaired FAT to a similar extent as 10 nM gp120. The local concentration of gp120 around DRG neurons has not been directly measured but is likely higher than plasma concentrations due to the close proximity of infected cells binding to extracellular matrix components or local glial swelling (Nath, 2002; Krathwohl and Kaiser, 2004). An important point is that DSP takes decades to develop, although subclinical signs are commonly manifested in HIV patients. Thus, smaller amounts of gp120 internalized by DRG neurons could gradually compromise neuronal function over decades by compromising FAT in DRG neurons.
Collectively, results from experiments here revealed a novel mechanism of neurotoxicity for gp120. Gp120 is internalized by neurons, then localized and transported in their axons (Berth et al., 2015). Intra-axonal gp120 inhibits FAT by affecting regulatory signaling cascades. As impaired FAT promotes axonal pathology, observations here are consistent with the dying-back neurodegeneration seen in DSP. Indeed, pharmacological inhibition of the signaling pathway through which gp120 inhibits FAT in isolated axoplasm was sufficient to prevent neuritic abnormalities in mammalian DRG neurons. This direct effect of gp120 does not exclude the possibility of indirect actions, and it is likely that multiple actions of gp120 synergize to harm DRG neurons.For example, the activation of CXCR4 by gp120 that leads to p38 MAPK activation might synergize with p38 MAPK activation caused by intra-axonal gp120. Additionally, TNF-α has been strongly implicated in the inflammatory response in the indirect pathway for DSP (Herzberg and Sagen, 2001; Keswani et al., 2003; Zheng et al., 2011). Intriguingly, TNF-α activates the NF-κB pathway through Tak1, which is the same pathway that we have found activated by intra-axonal gp120. These multiple mechanisms leading to activation of Tak1 could very well synergize with each other. Regardless, we propose that impairment of FAT by gp120 is a critical step in the pathway to the development of DSP, making DSP another member of the class of neurological diseases that can be considered dysferopathies. Promoting normal regulation of FAT may represent a promising avenue for therapeutic interventions for DSP.
Berth SH, Mesnard-Hoaglin N, Wang B, Kim H, Song Y, Sapar M, Morfini G, Brady ST. HIV Glycoprotein Gp120 Impairs Fast Axonal Transport by Activating Tak1 Signaling Pathways. ASN Neuro. 2016 Nov 20;8(6):1759091416679073. doi: 10.1177/1759091416679073. PMID: 27872270; PMCID: PMC5119683.
This does not mean that COVID-19 or experimental gene agent sourced gp120 can induce the same pathologies, as the glycoprotein is an insert in spike S1 as discussed previously and different strains of gp120 exist.
But it is an area where more research is urgently needed to exclude the possibility or so that appropriate diagnosis and treatments can be administered or developed if needed.
The mole is a measure of quantity. 100 picomoles is 1.0×10-10 mol, ie axonal quantities comparable to the efficacious concentration of some nerve agents.
The picomole quantities of gp120 that can inhibit FAT via signalling cascades and the long latencies involved further emphasise the importance of investigating the pathology now. Its not something you would want to confirm many years later when irreversible neurological damage has already been caused to a large number of individuals.
Therapeutics
Hydroxychloroquine
To finish with some good news, how do we limit damage caused by disrupting the MAPK pathways, notibly p38? Hydroxychloroquine is one answer and it's a great one:
Molecular Insights into the MAPK Cascade during Viral Infection: Potential Crosstalk between HCQ and HCQ Analogues (2020)
“Interestingly, prior data had indicated that the HCQ/CQ could influence the MAPK cascade. The main aim of this review is to address molecular mechanisms, beyond drugs, that can be helpful against viral infection for this and future pandemics. We will highlight (1) the contribution of the MAPK cascade in viral infection and (2) the possible use of MAPK inhibitors in curbing viral infections, alone or in combination with HCQ and quinoline analogues. We are convinced that understanding the molecular patterns of viral infections will be critical for new therapeutical approaches to control this and other severe diseases.”
Figure 1
The MAPK pathway: activators and inhibitors. This picture summarizes the main components of the MAPK pathways. Upon membrane receptor activation, or viral infection [3], Ras family GTPases trigger signaling cascades, leading to the activation of ERK, JNK, and p38, which in turn regulate transcription of a plethora of genes involved in many cell functions, including regulation of viral infection/replication. Inhibitors of specific MAPK proteins, discussed in this review, are shown: U0126 for MEK1, SB203580 for p38, and PD 98059 for ERK1/2. As discussed in the text, CQ and HCQ have the ability to interfere with the pathway at the level of Raf or, downstream, on ERK1/2 and p38 activation.
Protective effect of N-acetylcysteine on cellular energy depletion in a non-septic shock model induced by zymosan in the rat (1999)
Abstract
Recently, it was proposed that zymosan, a nonbacterial agent, causes cellular injury by inducing the production of peroxynitrite and consequent poly-(ADP-ribose) synthetase (PARS activation). Here we investigated whether in vivo N-acetylcysteine treatment inhibits cellular injury in macrophages collected from rats subjected to zymosan-induced shock. Macrophages harvested from the peritoneal cavity exhibited a significant production of peroxynitrite, as measured by the oxidation of the fluorescent dye dihydrorhodamine 123, and by nitrotyrosine. Furthermore, zymosan-induced shock caused a suppression of macrophage mitochondrial respiration, DNA strand breakage, and reduction of cellular levels of NAD+. In vivo treatment with N-acetylcysteine (40, 20, and 10 mg/kg, intraperitoneally, 1 and 6 h after zymosan) significantly reduced in a dose-dependent manner peroxynitrite formation and prevented the appearance of DNA damage, the decrease in mitochondrial respiration, and the loss of cellular levels of NAD+. Our study supports the view that the antioxidant and anti-inflammatory effect of N-acetylcysteine is also correlated with the inhibition of peroxynitrite production. In conclusion, N-acetylcysteine may be a novel pharmacological approach to prevent cell injury in inflammation.
Behavioral Evidence for a Tau and HIV-gp120 Interaction
After posting in January ‘22 another paper was published in May that provided further evidence of the contribution to neurological dysfunction by gp120’s interaction with the neuronal cytoskeleton.
Vijayan et al conducted an in vivo study using transgenic Tau mice and wild type mice as a control. This may help to explain the increased numbers of diagnoses of AD and deteriorations of pre-existing conditions since 2020, and especially since early 2021.
Tau P301L
Neuropathology
Tau hyperphosphorylation and conformational changes in the brain parenchyma start around seven months. Tangle-like pathology is mainly observed in the brain stem and spinal cord, and to a lesser extent in the midbrain and cerebral cortex (Terwel et al., 2005). Age-dependent increase in total tau measured in the cerebral spinal fluid (personal communication, reMYND).
Other Phenotypes
Premature death, attributed to hindbrain tauopathy, generally occurs between eight and 12 months (Dutschmann et al., 2010). Premature death is preceded by weight loss, hyperkyphosis, reduced activity, and upper airway dysfunction. At a young age (eight to ten weeks), Tau P301L mice have enhanced long-term potentiation in the dentate gyrus. This is before hyperphosphorylation and tauopathy are apparent (Boekhoorn et al., 2006), and may be due to acclerated dendritic spine maturation (Kremer et al., 2011).
Modification Details
These transgenic mice overexpress human tau, specifically the 4R/2N isoform, bearing the P301L mutation under the control of the neuron-specific murine Thy1 promoter (Terwel et al., 2005).
Despite successful virologic control with combination antiretroviral therapy (cART), about half of people living with the human immunodeficiency virus-1 (HIV) develop an HIV-associated neurocognitive disorder (HAND). It is estimated that 50% of individuals who are HIV-positive in the United States are aged 50 years or older. Therefore, a new challenge looms as individuals living with HIV increase in age. There is concern that Alzheimer’s disease (AD) may become prevalent with an earlier onset of cognitive decline in people living with HIV (PLWH). Clinical data studies reported the presence of AD biomarkers in PLWH. However, the functional significance of the interaction between HIV or HIV viral proteins and AD biomarkers is still not well studied. The main goal of the present study is to address this knowledge gap by determining if the HIV envelope glycoprotein 120 (HIV-gp120) can affect the cognitive functions in the Tau mouse AD model. Male Tau and age-matched, wild-type (WT) control mice were treated intracerebroventricularly (ICV) with HIV-gp120. The animals were evaluated for cognitive function using a Y-maze. We found that HIV-gp120 altered cognitive function in Tau mice. Notably, HIV-gp120 was able to promote a cognitive decline in transgenic Tau (P301L) mice compared to the control (HIV-gp120 and WT). We provide the first in vivo evidence of a cognitive interaction between an HIV viral protein and Tau mice.
Keywords: human immunodeficiency virus-1 (HIV), combination antiretroviral therapy (cART), HIV-associated neurocognitive disorder (HAND), intracerebroventricularly (ICV), Alzheimer’s disease (AD), central nervous system (CNS), cerebrospinal fluid (CSF), glycoprotein 120 (gp-120)
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder and one of the most common causes of neurodegenerative dementia in the elderly. It is characterized by a progressive impairment of cognitive functions. Currently, over 50 million people worldwide live with AD-related dementia, and this number is expected to increase to 131.5 million by 2050 [1,2]. The disease is neuropathologically characterized by the deposition of abnormal proteins, resulting in the formation of extracellular senile plaques and intracellular neurofibrillary tangles (NFTs). The senile plaques contain primarily neurotoxic amyloid-β (Aβ) whereas NFTs consist of abnormal hyperphosphorylated Tau aggregates. Phosphorylated Tau disrupts the microtubule assembly, leading to defective axonal transport, ultimately leading to synaptic starvation and neuronal death.
In 2018, an estimated 37.9 million people were living with human immunodeficiency virus-1 (HIV) [3,4]. HIV-associated neurocognitive disorder (HAND) is a common primary neurological disorder of the central nervous system, associated with an HIV infection. Despite successful virologic control with combination antiretroviral therapy (cART), about half of people living with HIV (PLWH) develop HAND. It is estimated that 50% of the U.S. HIV-positive population is aged 50 years or older. Therefore, a new challenge looms as individuals living with an HIV infection age. There is concern that AD may become prevalent, with an earlier onset of cognitive decline in PLWH.
Clinical data derived mostly from case studies reported the presence of AD biomarkers in PLWH [5,6,7]. For example, a virally-suppressed female case (52 years old) demonstrated AD neuropathology and age-related Tau astrogliopathy in the hippocampus [5]. Consistent with AD, the 52-year-old demonstrated severe learning, memory, and daily function impairments [5]. AD biomarkers (e.g., Aβ1–42, Tau) are found in the cerebrospinal fluid (CSF) of HAND patients [8].
Preclinical data support a potential interaction between HIV viral proteins, such as HIV envelope glycoprotein 120 (HIV-gp120), and AD biomarkers. For example, increased Tau phosphorylation has been reported in HIV-gp120 transgenic mice [9]. The treatment of primary hippocampal cell cultures with recombinant HIV-gp120 promoted Aβ1–42 secretion [10]. Aβ deposition was accelerated in HIV-gp120/APP/PS1 mice compared with APP/PS1 mice in the dentate gyrus [11,12,13,14,15].
Despite this evidence, the functional significance of HIV or HIV viral proteins and AD interaction is still poorly understood. The main goal of the present study is to address this knowledge gap by determining if HIV-gp120 can affect the cognitive function in the transgenic Tau mouse model.
2.1. HIV-gp120 Promotes the Onset of Cognitive Deficit of Tau Mice
We used 4-month-old, an age prior to the onset of cognitive deficit, male Tau mice [16]. The HIV-gp120 dose we used does not produce cognitive decline by itself. Therefore, any effect on cognition during this testing period was the result of HIV-gp120 and Tau interaction and not their individual effects.
In light of our research, we are reporting two key findings. First, HIV-gp120 alters the cognitive function of Tau mice. Second, Mdivi1 prevents the cognitive interaction between HIV-gp120 and Tau mice.
The brain is one of the first sites targeted by HIV. This virus enters the brain early in the disease process and then continues to produce central nervous system (CNS) dysfunction as the disease progresses. HIV enters target cells by binding its HIV envelope HIV-gp120. This viral protein produces several neurobehavioral effects in rodents that are characteristic of HIV/AIDS [19,20,21,22]. We used the Tau AD mouse model to determine any behavioral interaction with HIV-gp120. Tau transgenic mice were generated with a human Tau P301L mutation [23]. Signs of cognitive impairments were detected at 5 months [16]. Recently, we found these effects became more dramatically impaired as the mice aged [24]. Here, we injected HIV-gp120 directly into the brain and determined its effect on cognitive function in Tau mice. We found that HIV-gp120 altered cognitive function in Tau mice. Notably, HIV-gp120 was able to promote a cognitive decline in Tau mice compared to the control (HIV-gp120 and WT).
A role of abnormal mitochondrial dynamics has been found to be an early event in the AD disease process in studies using postmortem AD brains, AD cell cultures, and AD mouse models [25,26,27,28,29,30,31,32]. HIV altered neuronal mitochondrial fission/fusion although different patterns have been reported [33,34,35]. Exposure of human primary neurons to HIV-gp120 accelerates the balance of mitochondrial dynamics toward fission (fragmented mitochondria) and induces perinuclear aggregation of mitochondria and mitochondrial translocation of dynamin-related protein 1 (DRP1), leading to neuronal mitochondrial fragmentation [35]. In vitro, recombinant HIV-gp120 decreased the total and active dynamin 1-like levels in primary neurons [33]. Our data show that the pretreatment with a mitochondrial division inhibitor prevents the cognitive interaction between HIV-gp120 and Tau mice. Based on this background and our pharmacological data, it seems as though the HIV-gp120 promotes the onset of cognitive decline in Tau mice through a mechanism that involves the enhancement of mitochondrial fragmentation, and this deficit can be rescued with Mdivi-1. The focus on mitochondria does not negate the role of other mechanisms. For example, clinical [1,2,3,4] and preclinical [5] data have shown neuroinflammation in the brains of people with AD as well as in people living with HIV [6]. In AD, neuroinflammation, instead of being a mere bystander activated by emerging senile plaques and neurofibrillary tangles, contributes as much or more to the pathogenesis as do the plaques and tangles themselves. Tau can induce inflammation [7] and studies show that microglial activation contributes to Tau pathology during AD pathogenesis [8,9,10,11]. HIV-gp120 can also produce neuroinflammation [12]. Therefore, a synergistic effect on neuroinflammation may account for HIV-gp120, promoting cognitive decline in Tau mice.
In summary, the current studies provide in vivo evidence of a cognitive interaction between an HIV viral protein and an AD mouse model. These data suggest that HIV and AD are not independent conditions, but they can interact with each other when they are comorbid. These data suggest a contribution of mitochondria in the cognitive interaction between HIV-gp120 and Tau mice.
More:
Behavioral Evidence for a Tau and HIV-gp120 Interaction (2022)
Novel cleavage sites identified in SARS-CoV-2 spike protein
A big hat tip to Genervter Bürger and Charles Rixey for hi-lighting other papers. Along with viral origins, these further investigated how inserts can be cleaved off the spike protein, with the effect that the biological availability of gp120 is increased further still.
The far left sequences to CS-1 in (a) contain 4 variable loops. The exact composition of amino acids is less important for binding than the pattern of them and the presence of disulphide bonds:
Cleavage of the S protein by host proteases is essential for viral infection. Here, we discovered that the S protein contains two previously unidentified Cathepsin L (CTSL) cleavage sites (CS-1 and CS-2). Both sites are highly conserved among all known SARS-CoV-2 variants. Our structural studies revealed that CTSL cleavage promoted S to adopt receptor-binding domain (RBD) “up” activated conformations, facilitating receptor-binding and membrane fusion. We confirmed that CTSL cleavage is essential during infection of all emerged SARS-CoV-2 variants (including the recently emerged Omicron variant) by pseudovirus (PsV) infection experiment.
From: “Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies” (2022)
a Schematic illustration of the SARS-CoV-2 S glycoprotein in which the functional domains and cleavage sites are highlighted (NTD N-terminal domain, RBD receptor-binding domain, CTD C-terminal domain, FPPR fusion-peptide proximal region, HR1 heptad repeat 1, HR2 heptad repeat 2, TMD transmembrane domain). CTSL cleaves at CTSL cleavage site 1 (CS-1) and CTSL cleavage site 2 (CS-2). Furin cleaves at the S1/S2 site.b Overall structure of the SARS-CoV-2 S ectodomain (PDB entry: 6VXX). The CTSL cleavage sites, CS-1 and CS-2, are colored in red, while the furin cleavage site at S1/S2 is colored orange. The three protomers of the S trimer are colored in pink, purple and green. c Schematic illustration and SDS-PAGE analysis for the cleavage of SARS-CoV-2 S glycoprotein. The purified SARS-CoV-2 S protein ectodomain was incubated with different concentrations of CTSL (2–8 μg/mL). The N-terminal sequencing results for band 1 (60 kDa), band 2 (100 kDa), and band 3 (170 kDa) are also shown. All samples were subjected to SDS-PAGE, and bands were detected by Coomassie blue staining. d 4-Gly-mutant SARS-CoV-2 S cannot be cleaved by CTSL into the 60 kDa (band 1) and 100 kDa (band 2) fragments. WT S2p protein (1 μg) and mutant S protein (1 μg) were incubated with CTSL (8 μg/mL). All samples were subjected to SDS-PAGE, and bands were detected by silver staining. e The P1 and P2 residues in CS-1 and CS-2 were mutated to glycine, and the mutant SARS-CoV-2-2-S glycoprotein was named 4-Gly-mutant S. f Amino acid sequence alignment of residues around CS-1 and CS-2 in SARS-CoV-2 variants. P1 and P2 residues that are the same as the WT residues are highlighted in red. The symbol * indicates amino acid residues that are conserved among all tested sequences. VOC variant of concern, VOI variant of interest, VUM variant under monitoring. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies (2022)
Charles Rixey: BTW, here's a graphic I made which helps elucidate the magical journey that variable loop bits from HIV took to appear in variable N-loops on a completely unrelated coronavirus, and [as pointed out by jikky et al] sit on the absolute most exposed and most useful points possible on that completely unrelated cov, which magically perfectly works as a DC receptor.
Added 15th October ‘23:
New pre-proof further supports the gp120 hypothesis and the link to brain cancer cases
On 13th October Horowitz et al published a hypothesis that linked brain fog to SARS-CoV-2 induced astrocyte metabolic dysfunction and brain fog.
Reading time: novelette TL;DR: PET scans have revealed that mRNA shot induced heart damage is in fact very common. Post shot or infection induced chronic inflammation may persist for years, contributing to the symptoms of “long Covid” and with elevated cardiovascular & cancer risk. A majority of deaths recorded as “natural causes” were associated with…
Brain [18F]FDG-PET scans have revealed a glucose hypometabolic pattern in patients with long COVID. This hypometabolism might reflect primary astrocyte dysfunction. Astrocytes play a key role in regulating energy metabolism to support neuronal and synaptic activity, especially activity involving glutamate as the main neurotransmitter. Neuroinflammation is one of the purported mechanisms to explain brain damage caused by infection with SARS-CoV-2. Microglial activation can trigger reactive astrogliosis, contributing to neuroinflammatory changes. These changes can disturb glutamatergic homeostasis, ultimately leading to cognitive fatigue, which has been described in other clinical situations. We hypothesize that glutamatergic dysregulation related to astrocyte dysfunction could be the substrate of brain PET hypometabolism in long COVID patients with brain fog. Based on these elements, we propose that therapeutics targeting astrocytic glutamate regulation could help mitigate long COVID neurological manifestations.
Keywords
PET Long COVID Post-COVID condition PASC Glutamate Metabolism Brain fog Astrocyte
From: “Fig. 1. Hypometabolism detected with 18F[FDG]-PET in long COVID patients: putative astrocyte dysfunction and glutamatergic dysregulation. (A) 18F[FDG]-PET scan of a healthy subject. (B) 18F[FDG]-PET scan of a long COVID patient. Hypometabolic areas are indicated with arrows. (C) A diagram showing a healthy condition in which an astrocyte has a normal capacity to take up glutamate and recycle it to sustain glutamatergic neurotransmission, giving rise to normal glucose utilization and lactate production to ensure adequate neuronal energy supply. (D) A diagram showing a long COVID condition in which an astrocyte has reduced glutamate uptake capacity, leading to reduced glutamate recycling, lower glucose utilization (observed with 18F[FDG]-PET as hypometabolism) and lower lactate production, dysregulating glutamatergic neurotransmission and possibly endangering neuronal survival.”, source: https://ars.els-cdn.com/content/image/1-s2.0-S0306987723001822-gr1_lrg.jpg
From: “Brain fog in long COVID: A glutamatergic hypothesis with astrocyte dysfunction accounting for brain PET glucose hypometabolism” (13th Oct. ‘23)
Parallels with gp120. Metabolic dysfunction in astrocytes leads to the formation of advanced glycation end products (AGEs) which alters normal synaptic plasticity.
Changes to synaptic plasticity can cause synapses to weaken over time and reduce the number of neurotransmitter receptors. This affects how well neurons communicate with each other.
SMI-LD: Spatial memory impairments and learning disabilities.
…Our lab has demonstrated that the HIV-1 protein, gp120, promotes SMI-LD by altering mitochondrial functions and energy production. We have investigated cellular processes upstream of the mitochondrial functions and discovered that gp120 causes metabolic reprogramming.Effectively, the addition of gp120 protein to neuronal cells disrupted the glycolysis pathway at the pyruvate level. Looking for the players involved, we found that gp120 promotes increased expression of polypyrimidine tract binding protein 1 (PTBP1), causing the splicing of pyruvate kinase M (PKM) into PKM1 and PKM2. We have also shown that these events lead to the accumulation of advanced glycation end products (AGEs) and prevent the cleavage of pro-brain-derived neurotrophic factor (pro-BDNF) protein into mature brain-derived neurotrophic factor (BDNF). The accumulation of proBDNF results in signaling that increases the expression of the inducible cAMP early repressor (ICER) protein which then occupies the cAMP response element (CRE)-binding sites within the BDNF promoters II and IV, thus altering normal synaptic plasticity. We reversed these events by adding Tepp-46, which stabilizes the tetrameric form of PKM2. Therefore, we concluded that gp120 reprograms cellular metabolism, causing changes linked to disrupted memory in HIV-infected patients and that preventing the disruption of the metabolism presents a potential cure against HAND progression.
From: “FIGURE 1. gp120 alters mitochondrial maximal respiration. (A) A schematic of typical results from a Seahorse Mito Stress Test showing the interpretation of each measurement taken at different time points. (B) Measurement of oxygen consumption rate (OCR) in LUHMES cells untreated (dotted lane/red squares) or treated with 100 ng/ml of gp120 protein (solid lane/black circles) for 8 h using a Seahorse XFe96 analyzer and a Mito stress test kit from Seahorse. Oligomycin, FCCP, and rotenone/antimycin (dotted lines) were added at 23, 50, and 75 min, respectively. (C) Graph displaying the maximal respiration and spare capacity measured (pmol/min) in LUHMES untreated or treated with gp120. Values derived from the Seahorse analyzer and computed using Seahorse software were considered statistically significant (*p < 0.05).”; source: https://www.frontiersin.org/files/Articles/812887/fncel-16-812887-HTML/image_m/fncel-16-812887-g001.jpg
From: “Metabolic Reprogramming in HIV-Associated Neurocognitive Disorders” (March ‘22)
As a side note, more and more cases of brain cancer are being reported. I know first hand of cases with very rapid progression and a bad outcome within weeks.
Spike protein has been found to accumulate in the skull-meninges-brain axis:
Abstract
Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), has been associated mainly with a range of neurological symptoms, including brain fog and brain tissue loss, raising concerns about the virus’s acute and potential chronic impact on the central nervous system. In this study, we utilized mouse models and human post-mortem tissues to investigate the presence and distribution of the SARS-CoV-2 spike protein in the skull-meninges-brain axis. Our results revealed the accumulation of the spike protein in the skull marrow, brain meninges, and brain parenchyma. The injection of the spike protein alone caused cell death in the brain, highlighting a direct effect on brain tissue. Furthermore, we observed the presence of spike protein in the skull of deceased long after their COVID-19 infection, suggesting that the spike’s persistence may contribute to long-term neurological symptoms. The spike protein was associated with neutrophil-related pathways and dysregulation of the proteins involved in the PI3K-AKT as well as complement and coagulation pathway. Overall, our findings suggest that SARS-CoV-2 spike protein trafficking from CNS borders into the brain parenchyma and identified differentially regulated pathways may present insights into mechanisms underlying immediate and long-term consequences of SARS-CoV-2 and present diagnostic and therapeutic opportunities.
From: “Short SummaryThe accumulation of SARS-CoV-2 spike protein in the skull-meninges-brain axis presents potential molecular mechanisms and therapeutic targets for neurological complications in long-COVID-19 patients.”; source: https://www.biorxiv.org/content/10.1101/2023.04.04.535604v1
From: “SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-Brain Axis: Potential Implications for Long-Term Neurological Complications in post-COVID-19” (April ‘23)
Homologous Gp120 from cleaved Spike S1 is implicated as contributory factor:
Abstract
Patients infected with human immunodeficiency virus (HIV) are more prone to developing cancers, including glioblastomas (GBMs). The median survival for HIV positive GBM patients is significantly shorter than for those who are uninfected, despite the fact that they receive the same treatments. The nature of the GBM–HIV association remains poorly understood. In this study, we analyzed the effect of the HIV envelope glycoprotein gp120 on GBM cell proliferation. Specifically, we performed cell cycle, western blot, protein synthesis and metabolomics analysis as well as ATP production and oxygen consumption assays to evaluate proliferation and metabolic pathways in primary human glioma cell line, U87, A172 cells and in the HIVgp120tg/GL261 mouse model. Glioma cells treated with gp120 (100 ng/mL for 7–10 days) showed higher proliferation rates and upregulation in the expression of enolase 2, hexokinase and glyceraldehyde-3-phosphate dehydrogenase when compared to untreated cells. Furthermore, we detected an increase in the activity of pyruvate kinase and a higher glycolytic index in gp120 treated cells. Gp120 treated GBM cells also showed heightened lipid and protein synthesis. Overall, we demonstrate that in glioma cells, the HIV envelope glycoprotein promotes proliferation and activation of glycolysis resulting in increased protein and lipid synthesis.
Keywords: glioma, HIV, gp120, glycolysis
From: “Figure 1. gp120 stimulates viability and migration and reduces cell death in glioma cells. Experiments were performed for untreated glioma cells and cells continuously treated with gp120 for 10 days. U87 and A172 cell lines and 965 primary glioma cells were investigated. Live and dead cells were counted with the use of trypan blue staining. (A) Cell viability was evaluated as the total number of live cells. (B) The proportion of dead cells was evaluated as the percentage of the total number of cells. (C) Migration assays were performed using Transwell membranes for 5 h of migration toward 5% serum-containing medium in the lower compartment. The total number of cells that had migrated to the lower compartment was determined and is represented in the graph. (D) H&E staining of brain sections encompassing glioma tumor and quantification of tumor size in wild type (WT) and HIVgp120tg mice. (E) Survival analysis, performed for WT and HIVgp120tg mice following GL261 glioma cells implantation. Mean ± S.E. and significant differences from control (*) are shown (p < 0.05). Unpaired t-tests were used to determine the significance between groups. Comparison of survival curves was performed by use of long-rank Mantel-Cox test. Six repeated experiments (n = 6) were used for statistical analysis.”; source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6162763/
From: “HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell”; (Sept ‘18)
If you got this far thanks for sticking with it, a short test is to follow…
Addendum
How to BLAST your way to the truth about the origins of COVID-19
Further confirmation of the presence of GP120 inserts, including by BLAST analysis of the genome sequences, which you can do yourself to verify the findings:
How to BLAST your way to the truth about the origins of COVID-19 (2021)
Dr Ah Kahn Syed
Using BLAST is easy. I'm going to show you how easy and how to prove that SARS-Cov-2 is man-made.
I’ve been meaning to write this blog for ever. Well, at least since Prashant Pradhan (a wonderful, honest and brave genomics scientist) raised the possibility back in February 2020 that the SARS-Cov2 virus was man made. And we have seen multiple confirmatory pieces that the virus was made in a lab, one of the better ones here on zenodo and with its own cute video for non-Bayesian peeps here. As of writing this those links are still up which at 12 months is pretty good going for any article that dares challenge the drivel propagandised by our beloved “free press [sponsored by pharma]”.
Anyway, BLAST is the NCBI/NIH (aka US government) repository for genomic and proteomic sequences, amongst other things. It is where all genome scientists around the world deposit their sequences if they make a discovery. Its main function is to allow comparison of gene sequences and discovery of sequences that match one that you might have come across in your experiment. What’s a gene sequence? That’s easy. It’s a line of code, made up of any combination of 4 letters in a sequence. Remember the film GATTACA? If you haven’t watched it by now, you should - because it’s yet another dystopian movie that is now too close to home.
DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.
Thank you for posting this article. Thank you for sharing about Hydroxychloroquine. Not being able to STOP or slow down the progress of dementia and help the loved while they are loosing their ability to speak and reason is for many people one of the hardest experiences they will go through life. So again, thank you for sharing about Hydroxychloroquine.




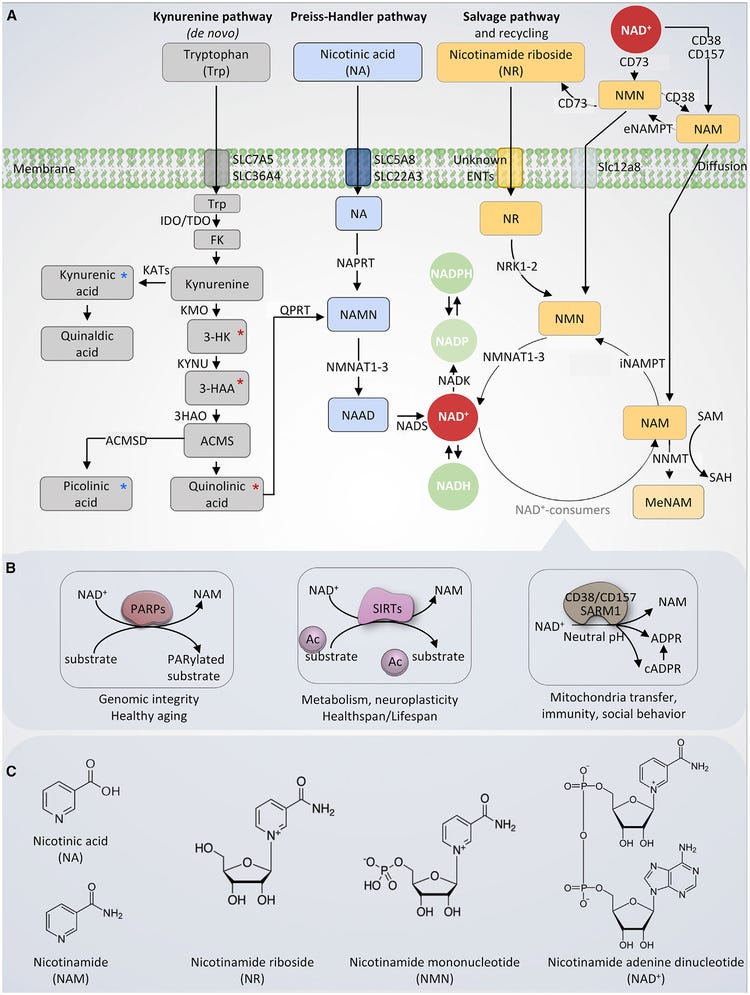


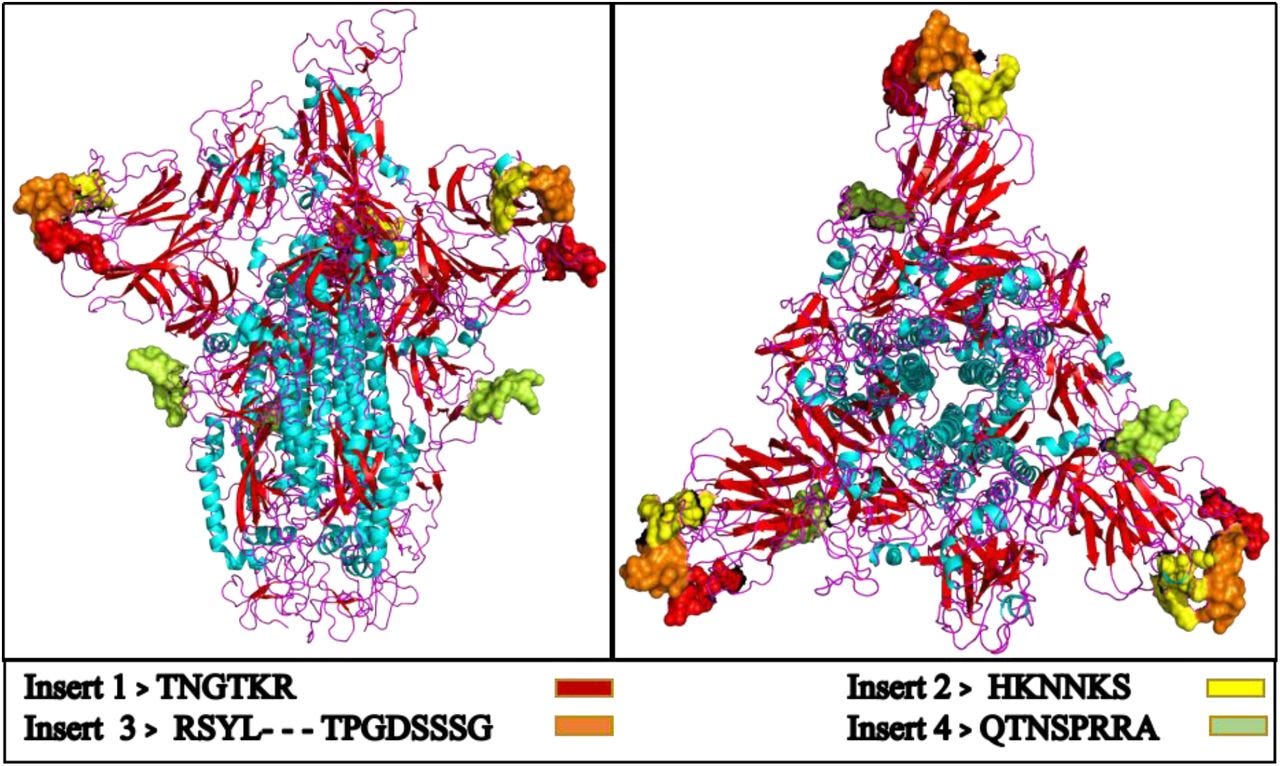



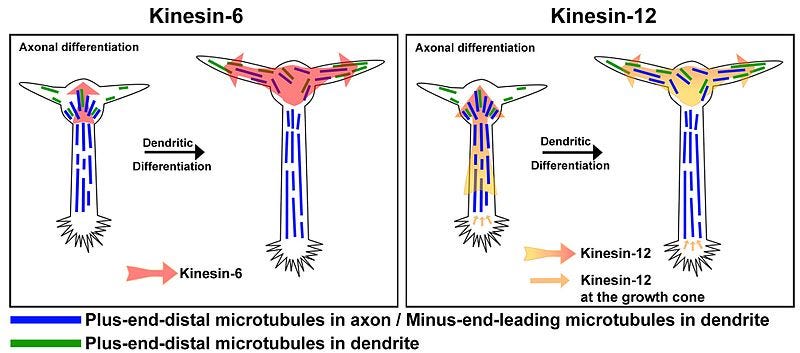

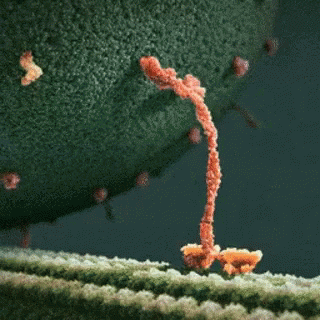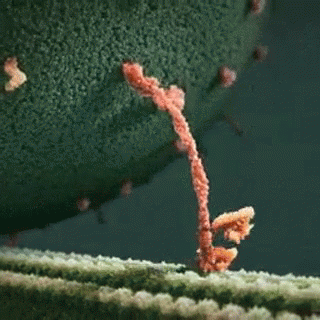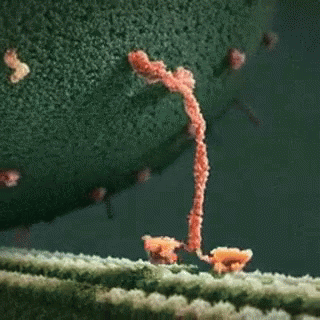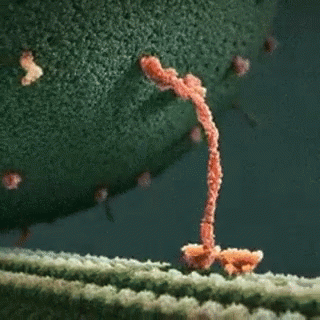
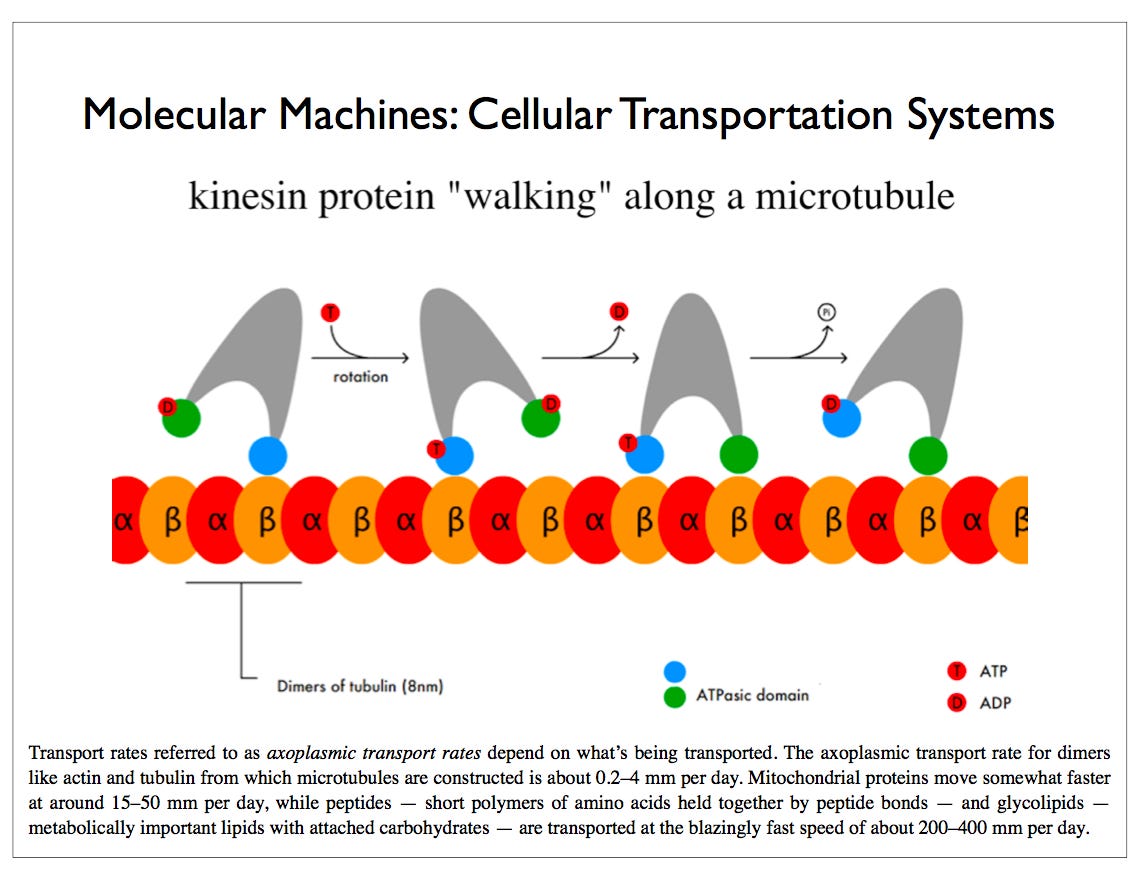

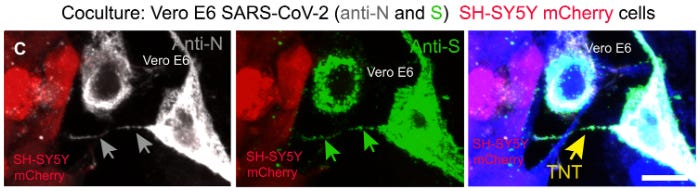

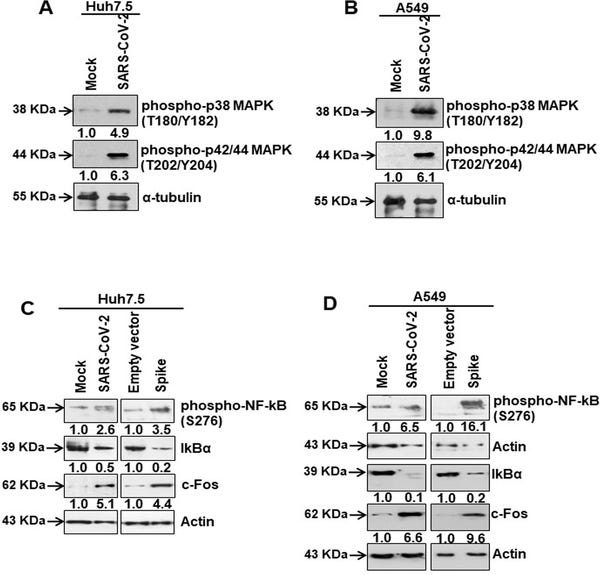
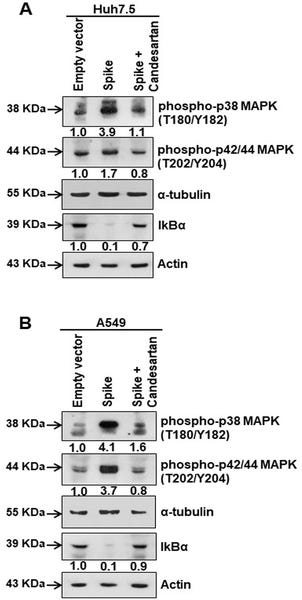
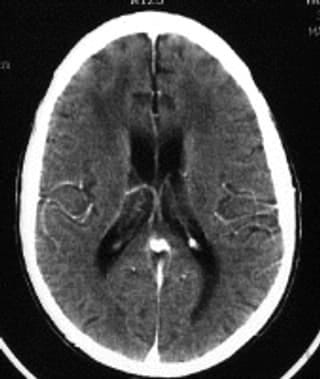




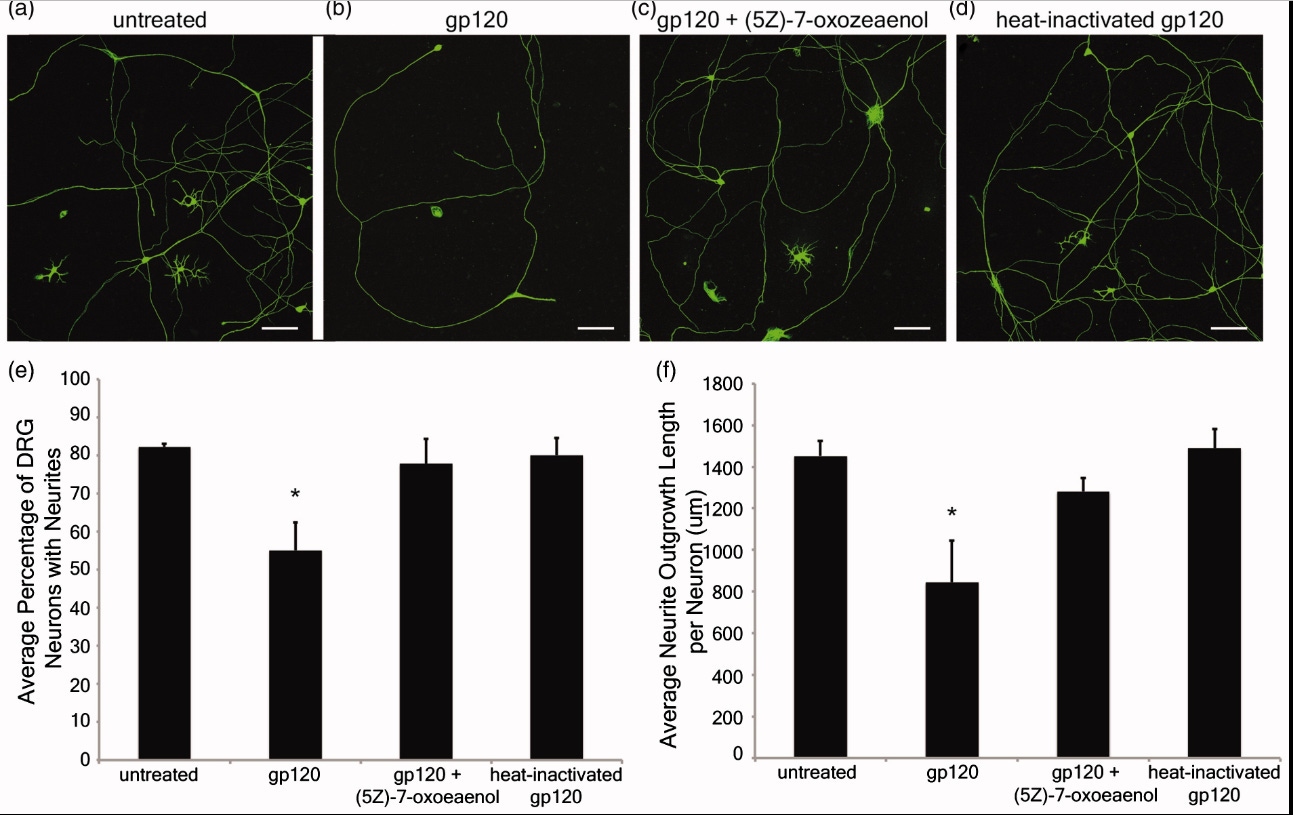



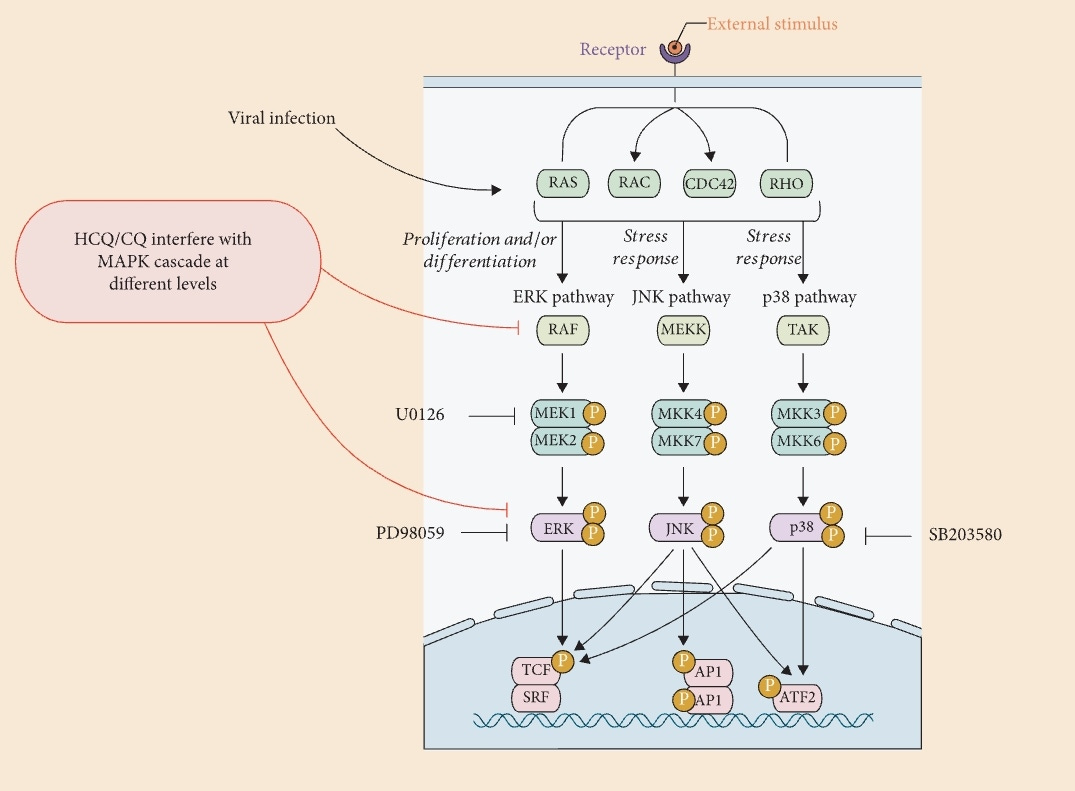



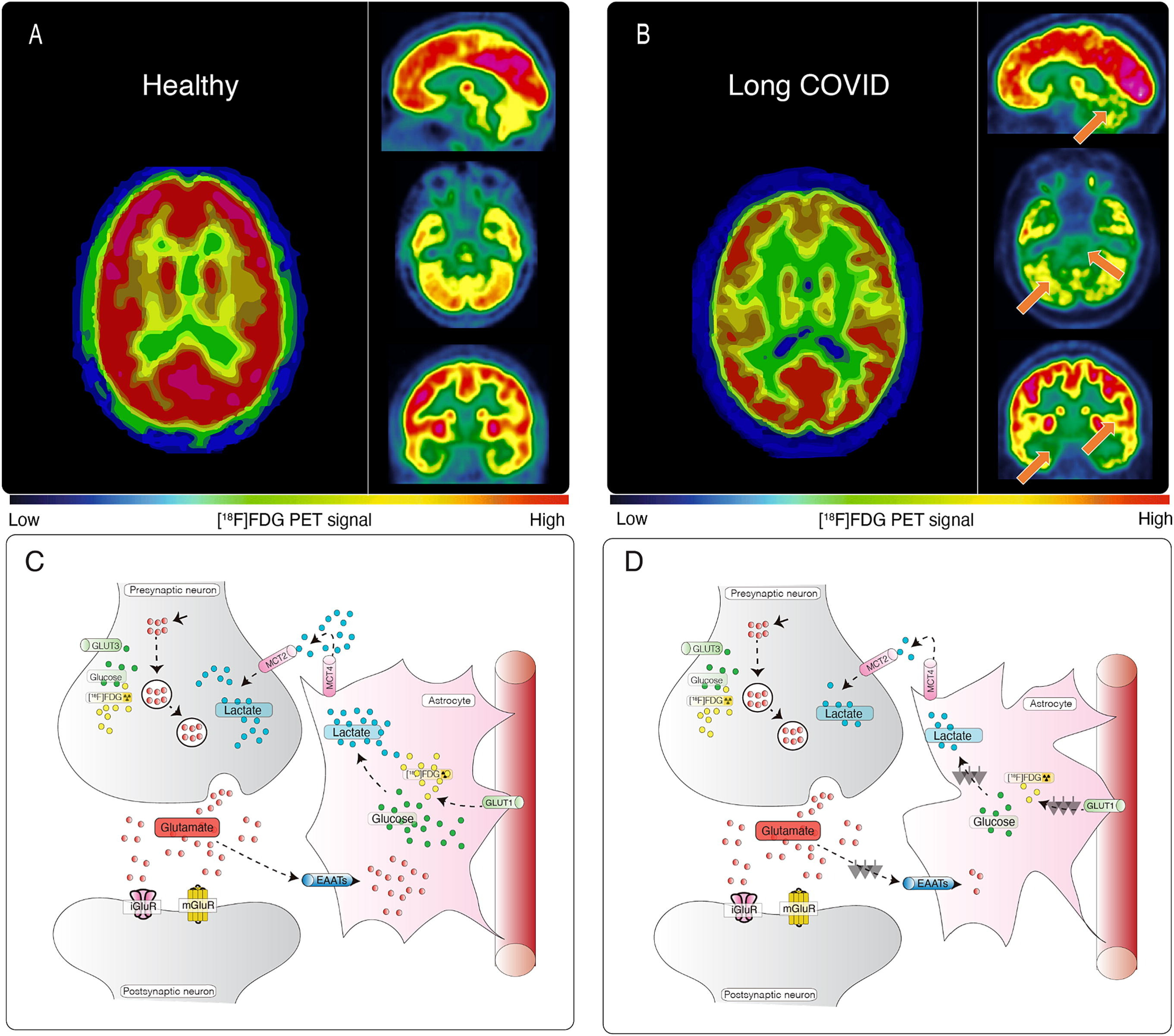
{kind=link}

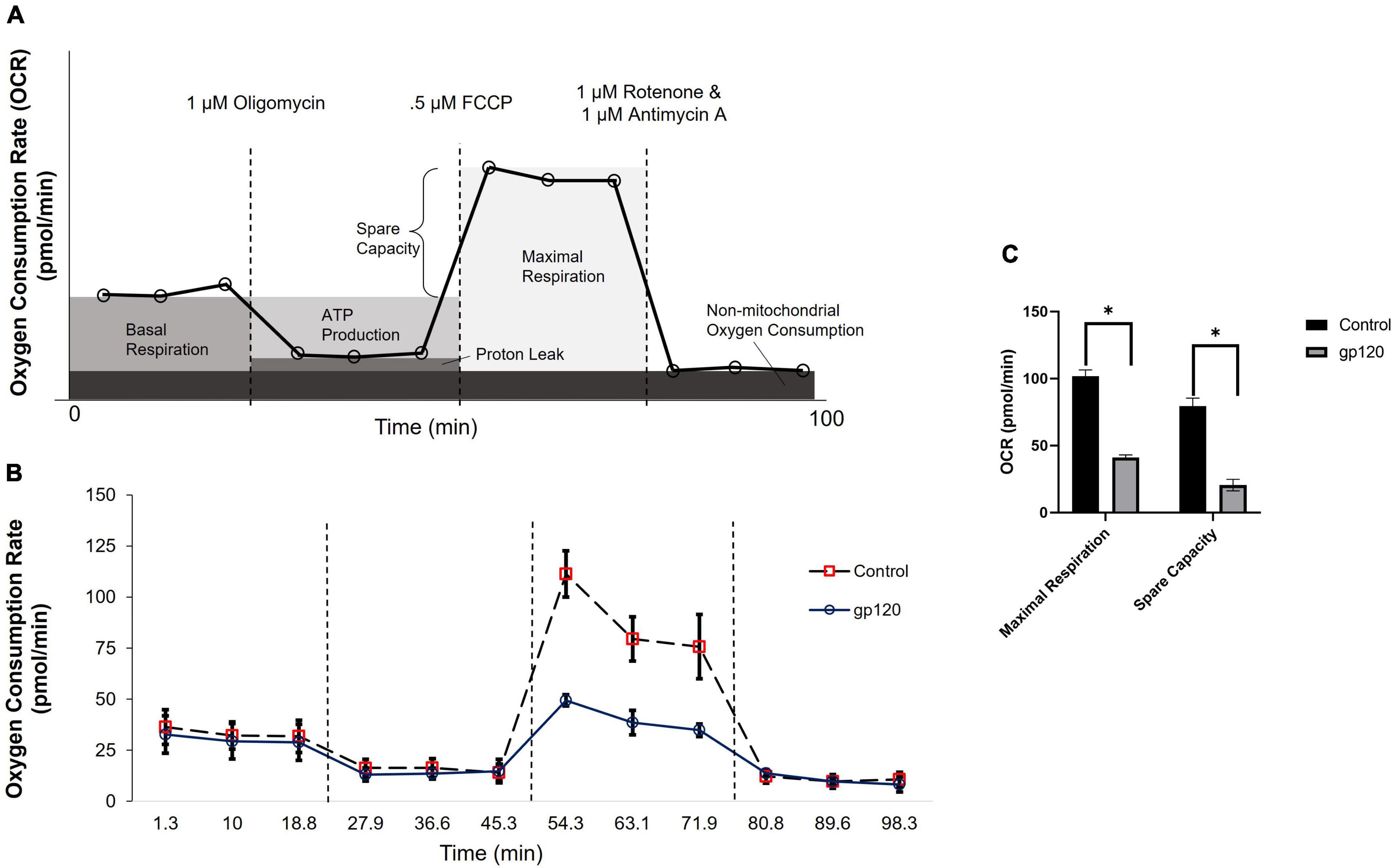{kind=link}


SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-
Brain Axis: Potential Implications for Long-Term Neurological
Complications in post-COVID-19 (2023)
https://www.biorxiv.org/content/10.1101/2023.04.04.535604v1
https://t.co/SfVOwSOl4G
Thank you for posting this article. Thank you for sharing about Hydroxychloroquine. Not being able to STOP or slow down the progress of dementia and help the loved while they are loosing their ability to speak and reason is for many people one of the hardest experiences they will go through life. So again, thank you for sharing about Hydroxychloroquine.