Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
In October 2021 Jiang & Mei published the ground breaking in vitro study “SARS–CoV–2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro” which provided strong experimental demonstrating that spike protein translocates to the nucleus1.
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS–CoV–2) has led to the coronavirus disease 2019 (COVID–19) pandemic, severely affecting public health and the global economy. Adaptive immunity plays a crucial role in fighting against SARS–CoV–2 infection and directly influences the clinical outcomes of patients. Clinical studies have indicated that patients with severe COVID–19 exhibit delayed and weak adaptive immune responses; however, the mechanism by which SARS–CoV–2 impedes adaptive immunity remains unclear. Here, by using an in vitro cell line, we report that the SARS–CoV–2 spike protein significantly inhibits DNA damage repair, which is required for effective V(D)J recombination in adaptive immunity. Mechanistically, we found that the spike protein localizes in the nucleus and inhibits DNA damage repair by impeding key DNA repair protein BRCA1 and 53BP1 recruitment to the damage site. Our findings reveal a potential molecular mechanism by which the spike protein might impede adaptive immunity and underscore the potential side effects of full-length spike-based vaccines.
Keywords: SARS–CoV–2, spike, DNA damage repair, V(D)J recombination, vaccine
As two critical host surveillance systems, the immune and DNA repair systems are the primary systems that higher organisms rely on for defense against diverse threats and tissue homeostasis. Emerging evidence indicates that these two systems are interdependent, especially during lymphocyte development and maturation [7]. As one of the major double-strand DNA break (DSB) repair pathways, non-homologous end joining (NHEJ) repair plays a critical role in lymphocyte–specific recombination–activating gene endonuclease (RAG) –mediated V(D)J recombination, which results in a highly diverse repertoire of antibodies in B cell and T cell receptors (TCRs) in T cells [8]. For example, loss of function of key DNA repair proteins such as ATM, DNA–PKcs, 53BP1, et al., leads to defects in the NHEJ repair which inhibit the production of functional B and T cells, leading to immunodeficiency [7,9,10,11].
To exclude the possibility that the spike protein inhibits DNA repair by promoting DNA repair protein degradation, we checked the expression of some essential DNA repair proteins in NHEJ and HR repair pathways and found that these DNA repair proteins were stable after spike protein overexpression (Figure 3C). To determine how the spike protein inhibits both NHEJ and HR repair pathways, we analyzed the recruitment of BRCA1 and 53BP1, which are the key checkpoint proteins for HR and NHEJ repair, respectively. We found that the spike protein markedly inhibited both BRCA1 and 53BP1 foci formation (Figure 3D–G). Together, these data show that the SARS–CoV–2 full–length spike protein inhibits DNA damage repair by hindering DNA repair protein recruitment.
Figure 3 Severe acute respiratory syndrome coronavirus 2 (SARS–CoV–2) spike protein impedes the recruitment of DNA damage repair checkpoint proteins. (A) Membrane fraction (MF), cytosolic fraction (CF), soluble nuclear fraction (SNF), and chromatin-bound fraction (CBF) from HEK293T cells transfected with SARS–CoV–2 spike protein were immunoblotted for His-tag spike and indicated proteins. (B) Left: Immunoblots of DNA damage marker γH2AX in empty vector (E.V)– and spike protein–expressing HEK293T cells after 10 Gy γ-irradiation. Right: corresponding quantification of immunoblots in left. The values represent the mean ± SD (n = 3). Statistical significance was determined using Student’s t-test. **** p < 0.0001. (C) Immunoblots of DNA damage repair related proteins in spike protein–expressing HEK293T cells. (D) Representative images of 53BP1 foci formation in E.V– and spike protein-expressing HEK293 cells exposed to 10 Gy γ–irradiation. Scale bar: 10 µm. (E) Quantitative analysis of 53BP1 foci per nucleus. The values represent the mean ± SEM, n = 50. (F) BRCA1 foci formation in empty vector- and spike protein-expressing HEK293 cells exposed to 10 Gy γ–irradiation. Scale bar: 10 µm. (G). Quantitative analysis of BRCA1 foci per nucleus. The values represent the mean ± SEM, n = 50. Statistical significance was determined using Student’s t-test. **** p < 0.0001.
4. Discussion
Our findings provide evidence of the spike protein hijacking the DNA damage repair machinery and adaptive immune machinery in vitro. We propose a potential mechanism by which spike proteins may impair adaptive immunity by inhibiting DNA damage repair. Although no evidence has been published that SARS–CoV–2 can infect thymocytes or bone marrow lymphoid cells, our in vitro V(D)J reporter assay shows that the spike protein intensely impeded V(D)J recombination. Consistent with our results, clinical observations also show that the risk of severe illness or death with COVID–19 increases with age, especially older adults who are at the highest risk [22]. This may be because SARS–CoV–2 spike proteins can weaken the DNA repair system of older people and consequently impede V(D)J recombination and adaptive immunity. In contrast, our data provide valuable details on the involvement of spike protein subunits in DNA damage repair, indicating that full–length spike–based vaccines may inhibit the recombination of V(D)J in B cells, which is also consistent with a recent study that a full–length spike–based vaccine induced lower antibody titers compared to the RBD–based vaccine [28]. This suggests that the use of antigenic epitopes of the spike as a SARS–CoV–2 vaccine might be safer and more efficacious than the full–length spike. Taken together, we identified one of the potentially important mechanisms of SARS–CoV–2 suppression of the host adaptive immune machinery. Furthermore, our findings also imply a potential side effect of the full–length spike–based vaccine. This work will improve the understanding of COVID–19 pathogenesis and provide new strategies for designing more efficient and safer vaccines.
p53, also known as Tumor protein P53, cellular tumor antigen p53 (UniProt name), or transformation-related protein 53 (TRP53) is a regulatory protein that is often mutated in human cancers. The p53 proteins (originally thought to be, and often spoken of as, a single protein) are crucial in vertebrates, where they prevent cancer formation.[5] As such, p53 has been described as "the guardian of the genome" because of its role in conserving stability by preventing genome mutation.[6] Hence TP53[note 1] is classified as a tumor suppressor gene.[7][8][9][10][11]
The TP53 gene is the most frequently mutated gene (>50%) in human cancer, indicating that the TP53 gene plays a crucial role in preventing cancer formation.[5] TP53 gene encodes proteins that bind to DNA and regulate gene expression to prevent mutations of the genome.[12] In addition to the full-length protein, the human TP53 gene encodes at least 15 protein isoforms.
Breast cancer type 1 susceptibility protein is a protein that in humans is encoded by the BRCA1 (/ˌbrækəˈwʌn/) gene.[5] Orthologs are common in other vertebrate species, whereas invertebrate genomes may encode a more distantly related gene.[6] BRCA1 is a human tumor suppressor gene[7][8] (also known as a caretaker gene) and is responsible for repairing DNA.[9]
BRCA1 and BRCA2 are unrelated proteins,[10] but both are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA, or destroy cells if DNA cannot be repaired. They are involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double-strand breaks.[11][12] If BRCA1 or BRCA2 itself is damaged by a BRCA mutation, damaged DNA is not repaired properly, and this increases the risk for breast cancer.[13][14] BRCA1 and BRCA2 have been described as "breast cancer susceptibility genes" and "breast cancer susceptibility proteins". The predominant allele has a normal, tumor suppressive function whereas high penetrance mutations in these genes cause a loss of tumor suppressive function which correlates with an increased risk of breast cancer.[15]
BRCA1 combines with other tumor suppressors, DNA damage sensors and signal transducers to form a large multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC).
The choice of words in the the closing statements of the Jiang & Mei paper crossed the line, orders came from above and despite lack of proof of fraud or poor experimental technique arms were twisted and the paper soon got retracted:
We are issuing this expression of concern in consultation with the publisher to fulfil their reporting obligation regarding the publication [1] mentioned above.
One of the authors has raised concerns regarding the methodology employed in the study, the conclusions drawn and the insufficient consideration of laboratory staff and resources.
In order to keep the highest scientific standards, an in-depth investigation is initiated by the responsible editors together with the journal’s editorial office in collaboration with the editorial board, and in accordance with the Committee on Publication Ethics (COPE) guidance. The article will be updated and any necessary corrections made at the conclusion of the investigation process.
Neither the authors nor the handling editor have responded to requests for comment from Retraction Watch about the specific concerns and what “insufficient consideration of laboratory staff and resources” means.
Indeed, what does this mean exactly?
Lack of paper towels?
No coffee left in the staff kitchen? That sort of thing?
We still don’t know because they won’t release the emails.
Arkmedic discusses the FOI requests:
Academic papers should only be retracted in the case of proven fraud. The whole future of academic science may well rest on the outcome of this investigation.
Jiang & Mei were later totally vindicated by this study by Sattar et al (26 January 2023):
“Nuclear translocation of spike mRNA and protein is a novel feature of SARS-CoV-2”4
Although the S protein is a surface transmembrane type 1 glycoprotein, it has been predicted to be translocated into the nucleus due to the novel nuclear localization signal (NLS) “PRRARSV,” which is absent from the S protein of other coronaviruses. Indeed, S proteins translocate into the nucleus in SARS-CoV-2-infected cells. S mRNAs also translocate into the nucleus. S mRNA colocalizes with S protein, aiding the nuclear translocation of S mRNA. While nuclear translocation of nucleoprotein (N) has been shown in many coronaviruses, the nuclear translocation of both S mRNA and S protein reveals a novel feature of SARS-CoV-2.
Figure 3. The nuclear translocation of S protein and S mRNA includes both the outer surface and inside of the nucleus. Separate slides (see Figure 2) were imaged under a Leica Stellaris confocal microscope (Leica) using a 63x oil objective. The images were then deconvolved using Huygen Essential deconvolution software (Scientific Volume Imaging). Using the surface rendering function of an image processing IMARIS software. (A) S mRNA (red) on the nuclear surface (top) and inside the nucleus (bottom). White arrows indicate S protein on the nuclear surface (top image) or inside the nucleus (bottom image). (B) S protein (green) on the nuclear surface (top image) and inside the nucleus (bottom image). White arrows indicate S protein on the nuclear surface (top image) or inside the nucleus (bottom image). (C) The total distribution of S mRNA and S protein in the cells. The data were obtained by combining multiple images from an independent experiment. (D) The total colocalization between S mRNA and S protein in the cells. The data were obtained by combining multiple images from an independent experiment.
The NLS referred to is the nuclear localization signal “PRRARSV”, which is a functional furin cleavage motif.
Additionally, one of the significant differences in the S protein sequences of SARS-CoV and SARS-CoV-2 is the pat7 NLS motif. Our results showed that transient expression of S protein by a plasmid containing the full-length S protein DNA sequence in both primary NHBE cells and A549 cell line lead its wider subcellular distribution including nucleus. Whether S protein expression by the current vaccine platforms causes suboptimal expression of S protein on the cell surface due to the NLS remains to be determined.
In conclusion, the SARS-CoV-2 S protein has a functional pat7 NLS “PRRARSV,” that results in one out of four S proteins translocating into the nucleus in infected cells. S Protein appears to shuttle S mRNA (possibly the genome) into the nucleus of the infected cells. Transient expression of full-length S protein in A549 cells reveals its nuclear translocation suggesting S protein’s nuclear translocation is NLS-driven and independent of interacting with another viral protein. Thus NLS of the S protein may contribute to the evasion of the host immune response and is a novel feature of SARS-CoV-2.
We now know by BLAST sequencing, which I’ve done myself, that synthetic mRNA gene agents do indeed contain the furin NLS sequence5. This is a problem in itself as furin is associated with carcinogenesis, and therapeutics are being developed just to target it.
This is not something you would knowingly choose to inject yourself with, to express furin systemically for days, weeks or months even6:
As a result of elevated furin levels in a mutated cell, enhanced TACE activity would see an increase in the secretion of TNFα thereby sustaining a localised inflammatory environment allowing for the development of carcinogenic cells. As furin activates MMP activity, these carcinogenic cells have the potential to become metastatic. This review investigates the role that furin plays in the activation of TACE and MMPs and the effect that this has on a skin cells exposed to UV radiation, as well as that its role in cancer cells which undergoes metastasis, and how an understanding of the role played by this proprotein convertase, may assist in the design of new inhibitors which have therapeutic potential.
Working with GRP
Which brings us back to present day. John Paul posted an excellent but disturbing Substack on the subject of COVID-19 induced hyperglycaemia as a byproduct of liver infection:
Spike protein interacts with a heat shock chaperone protein called GRP78. We know from the Pradhan et al paper7 (also later retracted) that spike protein contains 4 glycosylated protein inserts that are functionally a close match to envelope proteins on HIV, and once cleaved from S1 spike protein you have a protein of high biological availability and low mass, ideal for crossing the blood brain barrier or interacting with tumour suppressor proteins in the nucleus, for example.
Its therefore unsurprising that many spike protein related pathologies are almost interchangeable with gp120 related pathologies. Please note it is NOT HIV, but shares homology with some of the neurotoxic, cytotoxic and oncogenic envelope proteins.
There are different variants of gp120, but the pattern of amino acids used and the V3 loop formation is more important for receptor binding than the actual sequences involved, and even with HIV this is a dynamic process and difficult to measure8.
These novel findings put forward the first evidence that GRP78 is a key player in HIV-1 clade B and C neuropathogenic discrepancies and can be used as a novel target for immunotherapies.
Studies have shown that HIV-1 Tat and gp120 proteins are neurotoxic and have been suggested as one of the contributing factors of HIV-1 associated-dementia.
Moreover, the results presented here, show for the first time, that HIV-1 gp120 clade B protein induced the expression of a key ER-stress chaperone, GRP78. We hypothesized that the overexpression of GRP78 by HIV-1 gp120 clade B may protect astrocytoma cells from ER stress and oxidative injury that may lead to NeuroAIDS.9
Chemokine receptor type 4
I was particularly interested in spike protein binding to a chemokine receptor well known from HIV pathology called CXCR4 and, in a sense hoping I was very wrong as some painful conditions such as peripheral neuropathy (PN) and many other disorders could possibly be discounted if there was little to no binding potential.
For example, from “Internalization and Axonal Transport of the HIV Glycoprotein gp120” by Berth et al (2015)10:
Abstract
The HIV glycoprotein gp120, a neurotoxic HIV glycoprotein that is overproduced and shed by HIV-infected macrophages, is associated with neurological complications of HIV such as distal sensory polyneuropathy, but interactions of gp120 in the peripheral nervous system remain to be characterized. Here, we demonstrate internalization of extracellular gp120 in a manner partially independent of binding to its coreceptor CXCR4 by F11 neuroblastoma cells and cultured dorsal root ganglion neurons. Immunocytochemical and pharmacological experiments indicate that gp120 does not undergo trafficking through the endolysosomal pathway. Instead, gp120 is mainly internalized through lipid rafts in a cholesterol-dependent manner, with a minor fraction being internalized by fluid phase pinocytosis. Experiments using compartmentalized microfluidic chambers further indicate that, after internalization, endocytosed gp120 selectively undergoes retrograde but not anterograde axonal transport from axons to neuronal cell bodies. Collectively, these studies illuminate mechanisms of gp120 internalization and axonal transport in peripheral nervous system neurons, providing a novel framework for mechanisms for gp120 neurotoxicity.
Worse still, as spike protein or gp120 is quite resistant to proteases and removal by ubiquitination it may stay in neurones for decades, causing pain, prickling and tingling and loss of sensation in disorders such as PN. It can cause nerve disruption even at very low levels. Berth et al wrote a follow-up paper in 201611.
Ubiquitination is a tightly regulated, highly specific, and ATP-dependent biological process carried out by a complex cascade of enzymes. Ubiquitination is an essential player in protein homeostasis, serving to rapidly remove unwanted or damaged proteins.12
FAT is Fast Axonal Transport. I discussed this and related pathologies and pathways in great detail in the “brain fog” Substack, so won’t go into great detail again:
Sensory neuropathies are the most common neurological complication of HIV. Of these, distal sensory polyneuropathy (DSP) is directly caused by HIV infection and characterized by length-dependent axonal degeneration of dorsal root ganglion (DRG) neurons. Mechanisms for axonal degeneration in DSP remain unclear, but recent experiments revealed that the HIV glycoprotein gp120 is internalized and localized within axons of DRG neurons. Based on these findings, we investigated whether intra-axonal gp120 might impair fast axonal transport (FAT), a cellular process critical for appropriate maintenance of the axonal compartment. Significantly, we found that gp120 severely impaired both anterograde and retrograde FAT. Providing a mechanistic basis for these effects, pharmacological experiments revealed an involvement of various phosphotransferases in this toxic effect, including members of mitogen-activated protein kinase pathways (Tak-1, p38, and c-Jun N-terminal Kinase (JNK)), inhibitor of kappa-B-kinase 2 (IKK2), and PP1. Biochemical experiments and axonal outgrowth assays in cell lines and primary cultures extended these findings. Impairments in neurite outgrowth in DRG neurons by gp120 were rescued using a Tak-1 inhibitor, implicating a Tak-1 mitogen-activated protein kinase pathway in gp120 neurotoxicity. Taken together, these observations indicate that kinase-based impairments in FAT represent a novel mechanism underlying gp120 neurotoxicity consistent with the dying-back degeneration seen in DSP. Targeting gp120-based impairments in FAT with specific kinase inhibitors might provide a novel therapeutic strategy to prevent axonal degeneration in DSP.
Since the amount of free gp120 in plasma has been determined to be in the low picomolar to low nanomolar range (Gilbert et al., 1991; Oh et al., 1992), 100 pM gp120 was perfused to determine if a lower, more physiological relevant concentration might affect transport. In fact, 100 pM gp120 impaired FAT to a similar extent as 10 nM gp120. The local concentration of gp120 around DRG neurons has not been directly measured but is likely higher than plasma concentrations due to the close proximity of infected cells binding to extracellular matrix components or local glial swelling (Nath, 2002; Krathwohl and Kaiser, 2004). An important point is that DSP takes decades to develop, although subclinical signs are commonly manifested in HIV patients. Thus, smaller amounts of gp120 internalized by DRG neurons could gradually compromise neuronal function over decades by compromising FAT in DRG neurons.
CXCR4 speeds up the process but isn’t essential, according to the authors:
The assumption in these studies was that kinase activation and neurotoxic effects of gp120 were dependent upon binding to its coreceptor CXCR4, a GPCR that promotes a transient activation of kinase signaling cascades. Sustained activation of either JNK or P38 MAPK can affect FAT (Morfini et al., 2006; Morfini, You, et al., 2009; Bosco et al., 2010; Morfini et al., 2013), but transient activation is unlikely to significantly affect transport. However, elimination of the CXCR4 response with AMD3100 pretreatment unmasked a slower activation of p38 MAPK after 30 min of gp120 exposure.
How might this phosphorylation of p38 MAPK occur with no CXCR4 activation? Given that a portion of gp120 internalization in F11 cells was independent of CXCR4 binding (Berth et al., 2015), the delayed activation of p38 MAPK, corresponding to the timing of gp120 internalization, could be due to the activation of signaling cascades by intracellular gp120. This delayed activation of P38 MAPK would be masked by activation through CXCR4.
CXCR4 has roles in neurodevelopment and carcinogenesis amongst others13:
Function
CXCR-4 is an alpha-chemokine receptor specific for stromal-derived-factor-1 (SDF-1 also called CXCL12), a molecule endowed with potent chemotactic activity for lymphocytes. CXCR4 is one of several chemokine co-receptors that HIV can use to infect CD4+ T cells. HIV isolates that use CXCR4 are traditionally known as T-cell tropic isolates. Typically, these viruses are found late in infection. It is unclear as to whether the emergence of CXCR4-using HIV is a consequence or a cause of immunodeficiency.
…CXCR4 is present in newly generated neurons during embryogenesis and adult life where it plays a role in neuronal guidance. The levels of the receptor decrease as neurons mature. CXCR4 mutant mice have aberrant neuronal distribution. This has been implicated in disorders such as epilepsy.[11]
…While CXCR4's expression is low or absent in many healthy tissues, it was demonstrated to be expressed in over 23 types of cancer, including breast cancer, ovarian cancer, melanoma, and prostate cancer. Expression of this receptor in cancer cells has been linked to metastasis to tissues containing a high concentration of CXCL12, such as lungs, liver and bone marrow.[18][19] However, in breast cancer where SDF1/CXCL12 is also expressed by the cancer cells themselves along with CXCR4, CXCL12 expression is positively correlated with disease free (metastasis free) survival. CXCL12 (over-)expressing cancers might not sense the CXCL12 gradient released from the metastasis target tissues since the receptor, CXCR4, is saturated with the ligand produced in an autocrine manner.[20] Another explanation of this observation is provided by a study that shows the ability of CXCL12 (and CCL2) producing tumors to entrain neutrophils that inhibit seeding of tumor cells in the lung.[21]
Breast cancer type 1 susceptibility protein and other spike protein receptors
Whilst looking for research into spike protein to CXCR4 binding in support of the above as a working hypothesis I came across a paper I hadn’t seem before. This not only also vindicates the previously discussed papers but it proved to be something of a bombshell in itself and somehow managed to escape the censors, perhaps because it only mentions the word “\/ a c c i n e” once.
It is peer reviewed, published in 2020 by Masimani et al and only available in full as a pdf, UNO:
“SARS-CoV-2 Kerala Isolate Spike Protein Induces Cancer Proliferating Markers for Lung and Breast Cancer: An In Silico Approach”14.
NB Kerala is a state on India's tropical Malabar Coast, and was one of the first areas to sequence the SARS-CoV2 genome from clinical isolates and published the findings in May 202015.
In brief, the researchers used a program called “ClusPro”16 to simulate protein to protein docking and “Patchdock“17 and “Firedock”18 for scoring information.
From their analysis they concluded that the elevated risk of spike protein binding to a range of peptides and receptors linked to cancer pathways puts both the cancer patient and non-cancerous individual at increased risk of cancer acceleration and induction.
Being an in silico analysis this doesn’t automatically translate into clinical cases, but the early signs are highly alarming. New onset cases across all age groups and aggressive recurrences are being reported daily. The decline in some types of cancer vs increased incidence of previously “rare” sporadic cancers since 2020 shows that lockdowns or lack of consultations can now be discounted as an explanation, that time has passed.
Cancer Mortality Update Week 21 2023 ➼ Cancer UCoD = 3.0% Excess (Spring Lull) ➼ Cancer MCoD = 6.9% Excess ➼ Ages 0-54 Cancers = 20.5% Excess ➼ PPI Cancer Treatment = 11.4% Excess This is NOT due to Covid, Long Covid, nor 'deferred screenings'. https://twitter.com/EthicalSkeptic/status/1666531781212880897?s=20
I will touch on breast & ovarian cancer latencies later, but suffice to say the earliest onset cancers include blood cancers such as lymphoma may become symptomatic in less than 2 years. Other types may take decades.
Thirty-five of the 44 cancers in this analysis were found to progress silently for 10 years or longer prior to detection representing 89% of the patients in this analysis. The results of this analysis differentiate cancer types that progress undetected over a period of years to identify new opportunities for early detection which increases the likelihood of successful treatment and alleviates the ever-growing cancer burden19.
Abstract: Background: Coronavirus disease (COVID 19) has been emerging as a major threat to humans all over the world. Severe Acute Respiratory Syndrome CoronaVirus 2 (nSARS-CoV-2) is the causative agent for the disease resulting in severe acute respiratory illness. Earlier, it took several years to come up with a vaccine or other sorts of treatments for viral diseases. But now with the advent of biotechnology and development of bio-informatic tools, the process has been accelerated. The WHO reports 39,806,488 affected cases and 1,112,208 deaths till today all over the world (17 Oct 2020). nSARS-CoV-2 has a greater influence on people with comorbidities mainly cancer.
Objective: The study herein attempts to understand the binding affinity of the spike protein of the novel coronavirus with the lung and breast cancer marker proteins by docking and ClusPro analysis.
Methods: The analysis was conducted in reference to hACE2 (human Angiotensin Converting Enzyme 2), the receptor of nSARS-CoV-2. Total 22 different marker proteins were analyzed using ClusPro.
Results: BRCA1 (Breast Cancer type 1 susceptibility protein) and CXCR4 (a chemokine receptor belonging to the G protein coupled receptor family) were found to exhibit higher binding affinities.-73.82 kcal/mol and -66.45 kcal/mol were the global energies they showed upon binding to S protein respectively.
Conclusion:Therefore, novel SARS-CoV-2 has a higher chance of inducing cancer in non-cancerous individuals and aids in cancer acceleration in cancer patients . This poses a threat to cancer patients and immunocompromised individuals. The study can be exploited to identify the optimal drug delivery system for novel SARS CoV2.
The possibility of inducing mutations in BRCA1 is extremely concerning as this may be considered a long term to permanent greatly elevated risk:
BRCA 1 is found to be a transcriptional cofactor that enhances Tat (Trans Activator of Transcription) dependent transcription of HIV [43]. BRCA1 is also found to regulate IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of innate inflammation and interferon-β responses [44]. Most of the breast cancer cases showed decreased concentration of BRCA 1. Spike protein of SARS-CoV-2 exhibits better binding with BRCA1. Due to this interaction, there is a possible chance of malfunctioning or mutation of BRCA1.Mutation of BRCA1 affects the process of DNA repair, leading to the production of abnormal proteins and abnormal cell proliferation. Chemokine receptor CXCR4 belongs to the G protein coupled receptor family; chemokine stromal derived factor 1 alpha (SDF 1 α) is its natural ligand. When the ligand binds to the receptor, the cytoskeleton rearrangement occurs in addition to cellcell/matrix adhesion [42]. The stromal cell-derived factor-1 plays a crucial role in cell migration involved in hematopoiesis. CXCR4 is generally over-expressed in breast cancer. Nearly 75% of triple negative breast cancers showed high expression of CXCR4 [22, 23]. In case of malfunctioning of CXCR4 due to interaction of spike protein, hematopoiesis gets affected leading to further complications (Figs. 8 and 9).
Fig. (7). Complex structure obtained from cluspro indicating the interaction of spike protein with marker proteins.
It would have been useful to have spike to p53 analysis too, but what we have here is devastating enough already.
Key:
1T15: Brca1 BRCT Domains.
30E8: CXCR4 chemokine receptor.
3QRF: FOXP3 dimer.
1LYW: CATHEPSIN D.
Fig. (8). Top three proteins obtained from Patchdock and Cluspro based on binding energy and weighted score for centre, respectively.
Fig. (9). Pathways involving the proteins considered and their impact on tumourigenesis.
CONCLUSION
It is evident that spike protein of SARS-CoV-2 has good affinity towards BRCA1, CXCR4, and certain other proteins too and importantly higher affinity than ACE for most of the proteins. Appropriate functioning of these proteins is necessary to maintain a healthy cell micro environment. Alterations in the expression level or functioning of these proteins due to the bound spike protein of novel SARS-CoV-2 may result in tumour acceleration in the basic stage cancer patients . From our results, it is clear that when cancer patients, especially of lung and breast cancer, are affected by SARSCoV-2; they have a higher probability of acceleration and progression of cancer since the lung and breast cancer proteins taken into consideration show high binding affinity with the spike protein in comparison with ACE2. Apart from this, it is important to note that SARS-CoV-2 may even lead to the death of advanced stage cancer patients, in particular of lung and breast cancer, since most of the marker proteins get over expressed in cancer cells. There is also a possible chance of tumour induction in non-cancerous individuals via altering the function of normal proteins. Therefore, it is obvious that the novel SARS-CoV-2 Kerala isolate has better potential to induce and proliferate tumours. Novel SARSCoV-2 can also be used as anti-cancer drug carrier.
I can’t really add anything to that, other than no conflict of interest was declared, financial or otherwise.
Persistent expression
Regardless of whether induced changes to BRCA1 are permanent or not due to mutations the duration of spike protein expression in lymph node germinal centres was found to be at least 15 weeks in a study by Turner et al of individuals who had received 2 doses of BNT162b220.
Once expressed, cleaved S1 spike protein or the gp120-containing proteins are free to circulate systemically via exosomal transport. Synthetic mRNA can also be trafficked to other cells in the same manner, with the exosome taking the place of the LNP delivery vehicle21.
“SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses” (2021)
By examining fine needle aspirates of draining axillary lymph nodes, we identified germinal centre B cells that bound S protein in all participants who were sampled after primary immunization. High frequencies of S-binding germinal centre B cells and plasmablasts were sustained in these draining lymph nodes for at least 12 weeks after the booster immunization. S-binding monoclonal antibodies derived from germinal centre B cells predominantly targeted the receptor-binding domain of the S protein, and fewer clones bound to the N-terminal domain or to epitopes shared with the S proteins of the human betacoronaviruses OC43 and HKU1. These latter cross-reactive B cell clones had higher levels of somatic hypermutation as compared to those that recognized only the SARS-CoV-2 S protein, which suggests a memory B cell origin. Our studies demonstrate that SARS-CoV-2 mRNA-based vaccination of humans induces a persistent germinal centre B cell response, which enables the generation of robust humoral immunity.
Fig. 1: Plasmablast and antibody response to SARS-CoV-2 immunization.a, Study design. Forty-one healthy adult volunteers (ages 28–73, 8 with a history of SARS-CoV-2 infection) were enrolled and received the BNT162b2 mRNA SARS-CoV-2 vaccine. Blood was collected before immunization, and at 3, 4, 5, 7 and 15 weeks after immunization. For 14 participants (ages 28–52, none with a history of SARS-CoV-2 infection), FNAs of ipsilateral axillary lymph nodes (LNs) were collected at 3, 4, 5, 7 and 15 weeks after immunization. b, c, ELISpot quantification of S-binding IgG- (b) and IgA- (c) secreting plasmablasts (PBs) in blood at baseline, and at 3, 4, 5 and 7 weeks after immunization in participants without (red) and with (black) a history of SARS-CoV-2 infection. d, Plasma IgG titres against SARS-CoV-2 S (left) and the RBD of S (right) measured by ELISA in participants without (red) and with (black) a history of SARS-CoV-2 infection at baseline, and at 3, 4, 5, 7 and 15 weeks after immunization. Dotted lines indicate limits of detection. Symbols at each time point in b–d represent one sample (n = 41). Results are from one experiment performed in duplicate.
Gp120 from HIV is oncogenic on its own by disruption of BRCA1, by induction of DNA damaging reactive oxygen species (ROS)22 and by induction of systemically elevated inflammation.
From “Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind” by Isaguliants et al (2021)23:
Simple Summary
People living with human immunodeficiency virus type 1 (HIV-1) (PLWH) are at increased risk of developing cancer despite successful antiretroviral therapy (ART). Here, authors suggest novel mechanism behind this phenomenon. HIV proteins, namely envelope protein gp120, accessory protein negative factor Nef, matrix protein p17, transactivator of transcription Tat and reverse transcriptase RT, are known to be oncogenic per se, to induce oxidative stress and to be released from the infected or expressing cells. These properties are proposed to underlie their capacity to affect bystander epithelial cells causing their malignant transformation, and to enhance tumorigenic potential of already transformed/cancer cells. HIV proteins can act alone or in collaboration with other known oncoproteins, specifically originating from the oncogenic human viruses such as human hepatitis B and C viruses, and human papilloma viruses of high carcinogenic risk, which cause the bulk of malignancies in people living with HIV-1 on ART.
Figure 1. The effect of HIV-1 on cells of the liver. Infection with HIV-1 and even exposition of hepatocytes (HP), hepatic stellate cells (HSC), Kupffer cells (KFC) to HIV-1 leads to production of reactive oxygen species (ROS) and induction of proinflammatory microenvironment, which in turn, promote/enhance replication of HBV, HCV, as well as HIV-1 itself, resulting in enhanced fibrosis, cirrhosis and development of hepatocellular carcinoma (HCC). Infections are depicted in red, secondary effects in dashed black, and events leading to tissue damage in ochre-colored lines.
2.2. Brain Cancer
PLWH are highly predisposed to developing brain cancer, including primary central nervous system lymphomas (PCNSL) and glioblastomas (GBM) [43,44]. In pre-ART era, brain tumors were registered in 10% of PLWH [43]. Prevalence of PCNSL in AIDS patients was 3600-fold greater than in the general population, reaching 12% in AIDS patients [44]. ART has dramatically reduced these rates, possibly due to the effect of protease inhibitors [45]. Still, the prevalence of brain tumors in PLWH appears to be higher than in general population: in USA; recorded prevalence of PCNSL in HIV-1 infected is 8.4% compared to <3.3% in the general US population [45,46] Also GBM occurs in PLWH (in various stages of HIV-1 infection) at a younger age and at a frequency 5.4- to 45-fold higher than in the general population [47]. Furthermore, the median survival rate in patients with GBM for PLWH is shorter than for HIV-1-negative patients receiving same treatment (an average of 8 compared to 14 months, respectively) [48].
The nature of the brain tumor-HIV-1 relationship is not fully understood. The majority of these tumors are central nervous system lymphomas but gliomas may develop as well. GBM tumors appear approximately three years after HIV-1 infection [43].
Glioma cells were shown to interact with the HIV-1 envelope protein gp120. This interaction promotes proliferation, migration, survival and stimulates glycolysis in glioma cell lines and tumor growth in animal models [127]. Increased glycolysis, also known as the Warburg effect characteristic of malignancy [128], results in increased protein and lipid synthesis, and promotes uncontrolled propagation (both proliferation and invasion) of tumor cells, as it provides them with glycolytic intermediary precursors required for the synthesis of DNA, proteins and lipids [127,129]. As Tat, gp120 inducesEMT and cell migration through the TGF-B1 and MAPK signaling pathways [115,130].
The epithelial–mesenchymal transition (EMT) is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells; these are multipotent stromal cells that can differentiate into a variety of cell types. EMT is essential for numerous developmental processes including mesoderm formation and neural tube formation. EMT has also been shown to occur in wound healing, in organ fibrosis and in the initiation of metastasis in cancer progression.24
Virally-induced cancer evolves over long periods of time in the context of a strongly oxidative microenvironment, on the background of chronic inflammation. Oxidative stress induced by chronic viral infection is one of the factors driving neoplastic transformation, ultimately leading to oncogenic mutations in many cellular signaling cascades that drive cell growth and proliferation [42,141]. Oxidative damage of chromosomal DNA and chronic immune-mediated inflammation are key features of HBV, HCV, HPV, and HIV-1 infections [42,141]. As we have earlier reviewed, numerous lines of evidence show that HIV-1 infection triggers pronounced oxidative stress in both laboratory models and the context of in vivo infection by deregulation of oxidative stress pathways with escalation of ROS production and by inducing mitochondrial dysfunction [141]. As a result, PLWH exhibit multiple markers of oxidative stress including DNA damage [134,142]. The enhancement of ROS production is mediated by the envelope protein Gp120, Tat, Nef, RT, and p17 [141,142,143,144,145,146].
6.2. Envelope Protein Gp120
Early findings indicated that gp120 increases free radical production from monocyte-derived macrophages (MDM) detected by spin-trapping methods, and that the spin trap adduct results from a reaction involving nitrogen oxide NO or its closely related oxidized derivatives [153]. We have earlier summarized a profound role of gp120 in the induction of oxidative stress [141], namely gp120 induces ROS production in cell lines of lymphoid origin, in the endothelial brain cells, astrocytes, neurons and microglia. In astrocytes, it enhances ROS production by several parallel mechanisms: via Fenton–Weiss–Haber reaction, NOX2 and NOX4, and cytochrome P450 2E1 (CYP2E1) [154,155]. The latter is mediated through the upregulation of CYP2E1 expression. In cancer (neuroblastoma) cells, gp120 induces proline oxidase that synthesized pyroline-5-carboxylate with concomitant generation of ROS (reviewed in [141]).
The effect of HIV-1/HIV-1 proteins on the cellular antioxidant defense system is controversial. They can both suppress and enhance antioxidant defense pathways [141]. Gp120 was shown to induce oxidative stress response. It up-regulates functional expression in cultured astrocytes of multidrug resistance protein 1 (Mrp1) which effluxes endogenous substrates glutathione and glutathione disulphide involved in cellular defense against oxidative stress [156]. It also upregulates the expression of nuclear factor erythroid derived 2-related factor 2 (Nrf2), a basic leucine zipper transcription factor which is known to regulate antioxidant defensive mechanisms) in human astrocytes, stimulating expression of key antioxidant defensive enzymes hemoxygenase (HO-1) and NAD(P)H dehydrogenase quinone1 (Nqo1) [157]. Pre-treatment of astrocytes with antioxidants or a specific calcium chelator BAPTA-AM, significantly blocks the upregulation of Nrf2, HO-1 and Nqo1 [157].
Look far left, and you see p17>Misfolded amyloidogenic assemblies>Amyloid Beta like peptides/metal ion.
Matrix protein p17 is a monoclonal antibody to both HIV and SARS-CoV-2 and can cross the blood-brain barrier25.
P17 is a SARS-CoV-2-specific Spike RBD-binding human monoclonal antibody identified using a solid-phase immunotube screening method (Yao et al., 2020). P17 binds to Spike protein trimers of B.1.1.7 without a reduction in affinity. Binding is significantly weakened in the case of B.1.351 Spike trimer, however. For a standard version of Spike protein, the antibody displayed cooperative binding with antibodies HB27, H014, and FC05 (Sun et al., 2021; Wang et al., 2020; Yao et al., 2020)26.
Figure 3. Suggestive mechanism of direct carcinogenic effects of HIV-1 proteins. HIV-1 infected cells express and release gp120, Tat, Nef, p17, RT, each capable of the induction of oxidative stress. (1) p17 may trigger the production of ROS through binding of redox active metal ions by its amyloidogenic assemblies [167]. (2) Nef may indirectly activate NADPH oxidase by activating the Vav/Rac/p21-activated kinase (PAK) signaling pathway involved in phagocytic NADPH oxidase activation and produce peroxynitrite [160]. (3) Tat induces oxidative action through several independent mechanisms via NADPH oxidase, spermine oxidase (SMO) induction and mitochondrial dysfunction [148]. (4) RT induces ROS through unknown mechanisms. There is ROS –dependent activation of the Twist [134], which regulates the expression of Nrf2, which stimulating the expression of antioxidant enzymes (HO1, Nqol1). In addition, the Twist regulates the expression of the Snail. Both transcription factors, Twist and Snail, are involved in epithelial to mesenchymal transduction (EMT). (5) Gp120 increases free radical production from monocyte-derived macrophages (MDM) inducing nitrogen oxide (NO). In astrocytes (AS), it enhances ROS production by several parallel mechanisms: via cytochrome P450 2E1 (CYP2E1), NOX2 and NOX4, and the Fenton-Weiss-Haber reaction. Multidrug resistance proteins (Mrps) involved in cellular defense against oxidative stress. Mrp4 (isoform of Mrp) involved in the regulation of ROS and it acts against ROS [156]. In neuroblastoma cells (NB) gp120 was shown to induce proline oxidase that produces pyroline-5-carboxylate with a concomitant generation of ROS [141]. Production of ROS, which damage of bystander cells inducing oxidative damage of DNA, proteins and lipids, apoptosis and inflammation. DNA damage drives genomic instability and promotes transformation of healthy cells, and propagation and dissemination of malignant cells [168]. Arrows indicate: purple arrows—secretion/entering the intercellular space; black arrows—relationships and interactions; red arrows—production of ROS; blue arrows—oxidative stress response. Text above arrows designates the processes leading to the production of ROS, and text below the arrows, forms of ROS.
7.2. Envelope Protein gp120
Envelope protein gp120 is known to be secreted by chronically infected cells [180,181], particularly from the intraepithelial immune cells even in presence of ART [98]. A subset of PLWH demonstrate persistent circulation in plasma of gp120 [182] and in saliva [98]. Moreover, gp120 was found in tissues of PLWH [183]. Brain cells can be directly exposed to gp120 secreted by infiltrated and infected microglia and astrocytes [127]. Gp120 is internalized by bystander cells through receptor-independent mechanisms [184]. Internalization of gp120 leads to the release of several proinflammatory, angiogenic, and lymphangiogenic factors from affected cells [185].
8. Conclusions
People living with human immunodeficiency virus receiving antiretroviral therapy are characterized by high prevalence of different forms of cancer affecting epithelial cells. HIV-1 does not infect epithelial cells, however both HIV virions and proteins were shown to be sequestered into epithelial cells and affect their functions. These proteins have three specific properties:
First, HIV proteins Tat, Nef, gp120, matrix protein p17, reverse transcriptase/RT induce oxidative stress with serious consequences in the form of DNA, protein and lipid damage, as well as changes in the intracellular signaling.
Second, Tat, Nef, gp120, matrix protein p17, RT have a direct carcinogenic potential as demonstrated in the series of in vitro experiments and experiments in the laboratory animals.
Third, Tat, Nef, gp120, matrix protein p17, reverse transcriptase/RT were shown to exit HIV expressing cells by different mechanisms, and, once present in the extracellular space, can be up-taken by innocent neighbor cells.
Sequestered/internalized by innocent bystander cells, these proteins modulate their metabolism, cell cycle progression, ability to differentiate, motility, redox balance (induce ROS) and genomic stability. Through this, they can trigger malignant transformation of normal cells. Another outcome is propagation (proliferation and dissemination) of already existing precancerous and cancer cells, and enhanced growth and metastatic activity of tumors expressing or exposed to HIV-1 proteins.
The authors declared no conflict of interest.
Cathepsin D
Cathepsin D is a protein that in humans is encoded by the CTSD gene.[5][6] This gene encodes a lysosomal aspartyl protease composed of a protein dimer of disulfide-linked heavy and light chains, both produced from a single protein precursor. Cathepsin D is an aspartic endo-protease that is ubiquitously distributed in lysosomes.[7] The main function of cathepsin D is to degrade proteins and activate precursors of bioactive proteins in pre-lysosomal compartments.[8] 27
Apart from being associated with breast cancer a study involving mice which had been genetically engineered to be deficient in cathepsin D found that they exhibited progressive atrophy of their intestinal mucosa and destruction of their lymphatic immune system.
Saftig et al “Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells” (1995):28
Abstract
Mice deficient for the major lysosomal aspartic proteinase cathepsin D, generated by gene targeting, develop normally during the first 2 weeks, stop thriving in the third week and die in a state of anorexia at day 26 +/- 1. An atrophy of the ileal mucosa first observed in the third week progresses towards widespread intestinal necroses accompanied by thromboemboli. Thymus and spleen undergo massive destruction with fulminant loss of T and B cells. Lysosomal bulk proteolysis is maintained. These results suggest, that vital functions of cathepsin D are exerted by limited proteolysis of proteins regulating cell growth and/or tissue homeostasis, while its contribution to bulk proteolysis in lysosomes appears to be non-critical.
Cathepsin D levels need to be maintained in a narrow band or pathologies can result, including immunodeficiencies or carcinogenesis:
Profound loss of lymphoid cells
Elevated numbers of apoptotic cells in the thymus were already detected in 14-day-old CTSD -/- mice parallel to the onset of the atrophy of the intestinal mucosa. In the immune system proliferation, differentiation and also elimination by programmed cell death of lymphocytes is tightly regulated by external as well as internal signalling molecules (Raff, 1992; Schwartz and Osborne, 1993; Williams and Smith, 1993). In CTSD -/- animals the ratio between proliferation and elimination of lymphocytes is disturbed in favour of elimination by apoptosis. The present study suggests, that CTSD is not essential for differentiation of T and B cells.
Overexpression of p53
Deficiency or mutated p53 is associated with many cancer types, especially breast, colorectal, liver, lung and ovarian29 but overexpression is also linked to various pathologies due to the induction of apoptosis of otherwise healthy cells.
Just like the cathepsins and other proteins it needs to be kept in balance.
Liu et al used a mouse model to study the neurotoxic effects of gp120 on cerebrocortical neurons.
“ASPP2 involvement in p53-mediated HIV-1 envelope glycoprotein gp120 neurotoxicity in mice cerebrocortical neurons” (2016):30
Abstract
The mechanisms behind HIV-1-associated neurocognitive disorders are still unclear. Apoptosis-stimulating protein 2 of p53 (ASPP2) is a damage-inducible p53-binding protein that stimulates p53-mediated apoptosis and transactivates proapoptotic and cell cycle regulatory genes. It has been reported that ASPP2 has a specific regulatory function in the death of retinal ganglion cells and the development of Alzheimer’s disease. In this study, we used p53 and ASPP2 knockout mice and primary cerebrocortical neuron culture to analyze the role of the interaction between ASPP2 with p53 in HIV-1 envelope glycoprotein gp120-induced neurotoxicity. The results showed that 10 ng/mL gp120 protein might stimulate p53 overexpression and translocation to the nucleus, and 30 ng/mL gp120 protein could stimulate both p53 and ASPP2 translocation to the nucleus, but only with p53 overexpression. The primary cultured neurons of p53−/−ASPP2+/− mice had a higher survival rate than p53−/− mice under gp120 protein stress. The interaction of ASPP2 with p53 induced by a high dose of gp120 stimulated Bax transcription and contributed to caspase-3 cleavage, and ASPP2-siRNA attenuated gp120 induced neuron death through inhibition of Bax expression. These results suggest that ASPP2 plays an important role in p53-mediated neuronal apoptosis under gp120 stress.
BRCA1 and Curcumin
Al-Yousef et al conducted in vitro studies to investigate how curcumin can suppress breast cancer cell lines.
This is just one of the cancer pathways which are targeted by curcumin31.
Take with piperine, see links below.
“Curcumin induces re-expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines” (2020)32.
Abstract
Restoration of normal DNA promoter methylation and expression states of cancer-related genes may be an option for the prevention as well as the treatment of several types of cancer. Constitutional promoter methylation of BRCA1 DNA repair associated (BRCA1) gene is linked with a high risk of developing breast and ovarian cancer. Furthermore, hypomethylation of the proto-oncogene γ synuclein (SNCG) is associated with the metastasis of breast and ovarian cancer and reduced disease-free survival (DFS). In the present study, we evaluated the potential of curcumin to re-express hypermethylated BRCA1 and to suppress hypomethylated SNCG in triple-negative breast cancer (TNBC) cell line HCC-38, the estrogen receptor-negative/progesterone receptor-negative (ER−/PR−) cell line UACC-3199, and the ER+/PR+ cell line T47D. The cells were treated with 5 and 10 µM curcumin for 6 days and with 5-aza-2′-deoxycytidine (5′-aza-CdR) for 48 h. Methylation-specific PCR and bisulfite pyrosequencing assays were used to assess DNA promoter methylation while gene expression levels were analyzed using quantitative real-time PCR and immunoblotting. We found that curcumin treatment restored BRCA1 gene expression by reducing the DNA promoter methylation level in HCC-38 and UACC-3199 cells and that it suppressed the expression of SNCG by inducing DNA promoter methylation in T47D cells. Notably, 5′-aza-CdR restored BRCA1 gene expression only in UACC-3199, and not in HCC-38 cells. Curcumin-induced hypomethylation of the BRCA1 promoter appears to be realized through the upregulation of the ten-eleven translocation 1 (TET1) gene, whereas curcumin-induced hypermethylation of SNCG may be realized through the upregulation of the DNA methyltransferase 3 (DNMT3) and the downregulation of TET1. Notably, miR-29b was found to be reversely expressed compared to TET1 in curcumin- and 5′-aza-CdR-treated cells, suggesting its involvement in the regulation of TET1. Overall, our results indicate that curcumin has an intrinsic dual function on DNA promoter methylation. We believe that curcumin may be considered a promising therapeutic option for treating TNBC patients in addition to preventing breast and ovarian cancer, particularly in cancer-free females harboring methylated BRCA1.
The “latency period” for a cancer is the time from inception of disease to clinical presentation.
Massachusetts State issued an informative guide: “RISK FACTOR INFORMATION FOR SELECTED CANCER TYPES - Breast Cancer.”33
Again I will highlight key passages, but by all means follow the link and download the full .rtf doc.
Breast Cancer
Breast cancer is the most frequently diagnosed cancer among women in both the United States and in Massachusetts. According to the North American Association of Central Cancer Registries, female breast cancer incidence in Massachusetts is the fifth highest among all states (Chen et al, 2000). Although during the 1980s breast cancer in the U.S. increased by about 4% per year, the incidence has leveled off to about 110.6 cases per 100,000 (ACS 2000). A similar trend occurred in Massachusetts and there was even a slight decrease in incidence (1%) between 1993 and 1997 (MCR 2000).
In the year 2005, approximately 211,240 women in the U.S. will be diagnosed with breast cancer (ACS 2005). Worldwide, female breast cancer incidence has increased, mainly among women in older age groups whose proportion of the population continues to increase as well (van Dijck, 1997). A woman’s risk for developing breast cancer can change over time due to many factors, some of which are dependent upon the well-established risk factors for breast cancer. These include increased age, an early age at menarche (menstruation) and/or late age at menopause, late age at first full-term pregnancy, family history of breast cancer, and high levels of estrogen. Other risk factors that may contribute to a woman’s risk include benign breast disease and lifestyle factors such as diet, body weight, lack of physical activity, consumption of alcohol, and exposure to cigarette smoke. Data on whether one’s risk may be affected by exposure to environmental chemicals or radiation remains inconclusive. However, studies are continuing to investigate these factors and their relationship to breast cancer.
According to recent studies, approximately 10% of breast cancers can be attributed to inherited mutations in breast cancer related genes. Most of these mutations occur in the BRCA1 and BRCA2 genes. Approximately 50% to 60% of women who inherit BRCA1 or BRCA2 gene mutations will develop breast cancer by the age of 70 (ACS 2001).
To date, no specific environmental factor, other than ionizing radiation, has been identified as a cause of breast cancer. The role of cigarette smoking in the development of breast cancer is unclear. Some studies suggest a relationship between passive smoking and increased risk for breast cancer; however, confirming this relationship has been difficult due to the lack of consistent results from studies investigating first-hand smoke exposure (Laden and Hunter, 1998).
Studies on exposure to high doses of ionizing radiation demonstrate a strong association with breast cancer risk. These studies have been conducted in atomic bomb survivors from Japan as well as patients that have been subjected to radiotherapy in treatments for other conditions (i.e., Hodgkin’s Disease, non-Hodgkin’s Lymphoma, tuberculosis, post-partum mastitis, and cervical cancer) (ACS 2001). However, it has not been shown that radiation exposures experienced by the general public or people living in areas of high radiation levels, from industrial accidents or nuclear activities, are related to an increase in breast cancer risk (Laden and Hunter, 1998). Investigations of electromagnetic field exposures in relation to breast cancer have been inconclusive as well.
Occupational exposures associated with increased risk for breast cancer have not been clearly identified. Experimental data suggests that exposure to certain organic solvents and other chemicals (e.g., benzene, trichloropropane, vinyl chloride, polycyclic aromatic hydrocarbons (PAHs)) causes the formation of breast tumors in animals and thus may contribute to such tumors in humans (Goldberg and Labreche, 1996). Particularly, a significantly elevated risk for breast cancer was found for young women employed in solvent-using industries (Hansen, 1999). Although risk for premenopausal breast cancer may be elevated in studies on the occupational exposure to a combination of chemicals, including benzene and PAHs, other studies on cigarette smoke (a source of both chemicals) and breast cancer have not shown an associated risk (Petralia et al, 1999). Hence, although study findings have yielded conflicting results, evidence does exist to warrant further investigation into the associations.
Other occupational and environmental exposures have been suggested to confer an increased risk for breast cancer in women, such as exposure to polychlorinated biphenyls (PCBs), chlorinated hydrocarbon pesticides (DDT and DDE), and other endocrine-disrupting chemicals. Because these compounds affect the body’s estrogen production and metabolism, they can contribute to the development and growth of breast tumors (Davis et al, 1997; Holford et al, 2000; Laden and Hunter, 1998). However, studies on this association have yielded inconsistent results and follow-up studies are ongoing to further investigate any causal relationship (Safe, 2000).
When considering a possible relationship between any exposure and the development of cancer, it is important to consider the latency period. Latency refers to the time between exposure to a causative factor and the development of the disease outcome, in this case breast cancer. It has been reported that there is an 8 to 15 year latency period for breast cancer (Petralia 1999; Aschengrau 1998; Lewis-Michl 1996). That means that if an environmental exposure were related to breast cancer, it may take 8 to 15 years after exposure to a causative factor for breast cancer to develop.
HIV-like DNA sequences, viral particles and breast cancer
Whilst researching for this Substack I found a paper from 1998 by Rakowicz-Szulczynska et al with somewhat striking conclusions in relation to breast cancer and HIV gene homology.
Are some cases of breast and other cancers a result of virus harvesting from simians to create HIV and other viruses decades ago, with years of gain of function work?34
“Human Immunodeficiency Virus Type 1-Like DNA Sequences and Immunoreactive Viral Particles with Unique Association with Breast Cancer” (1998)35
MAb RAK-BrI, which was developed against RAK antigens, also cross-reacts with HIV-1 antigens gp160 and gp120, which confirms the nonaccidental similarity of cancer and HIV-1 proteins.
ABSTRACT
RAK antigens p120, p42, and p25 exhibit molecular and immunological similarity to the proteins encoded by human immunodeficiency virus type 1 (HIV-1) and are expressed by 95% of breast and gynecological cancer cases in women and prostate cancer cases in men. The binding of an epitope-specific anti-HIV-1 gp120 monoclonal antibody (MAb) (amino acids 308 to 322) to cancer RAK antigens has been found to be inhibited by a peptide derived from variable loop V3 of HIV-1. Breast cancer DNAs of 40 patients were PCR amplified with HIV-1 gp41-derived primers, and all of the samples were found to be positive. The DNA fragments amplified in seven blindly selected breast cancer samples were sequenced. The breast cancer DNA sequences showed at least 90% homology to the HIV-1 gene for gp41. Antisense oligonucleotides complementary to the HIV-1-like sequences inhibited reverse transcriptase activity and inhibited the growth of breast cancer cells in vitro. Viral particles detected in breast cancer cell lines were strongly immunogold labeled with the anti-HIV-1 gp120 MAb. The results obtained strongly suggest that the long-postulated breast cancer virus may, in fact, be related to HIV-1.
Breast cancer affects 1 in every 8 to 10 women in the United States (10, 19, 32, 35, 36). Approximately 10% of breast cancer patients exhibit a genetic inheritance pattern, while the overwhelming majority of women develop breast cancer for an unpredictable reason (2, 6, 7, 14, 20, 23, 37, 43). Recent enthusiasm following the characterization of the BRCA1 and BRCA2 genes, which are associated with some inherited forms of breast and ovarian cancer, has been diminished by the fact that only 1 of 800 women carries the mutated BRCA gene, while at least 80 to 100 of 800 women will develop breast cancer (17, 21, 41, 42).
Involvement of a viral factor in the etiology of human breast cancer has been considered by several laboratories (1, 5, 9, 11–13, 18, 39). Special attention was focused on human DNA sequences with homology to mouse mammary tumor virus (MMTV) (1, 38, 40). Retrovirus-like particles with immunological similarity to MMTV proteins were found in human breast carcinoma cell lines (13), peripheral blood monocytes of breast cancer patients (11), pleural effusion fluids from breast adenocarcinoma patients (39), human breast cancer tissue (5, 9, 12, 39), and breast milk (18). No viral agent has been identified as a causative agent of breast cancer in humans.
Except for cervical cancer, which is associated predominantly with human papillomavirus and/or herpes simplex virus infection (3), the viral etiology of other types of female reproductive tract cancer remains unverified. Recent studies on herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma (4, 22, 25, 33) strongly suggest that the role of retroviruses in human cancer was underestimated for a long time.
We have recently identified a new class of breast and gynecological cancer markers, which have been named Rakowicz markers or, briefly, RAK markers (25–30). RAK antigens p120, p42, and p25 express epitopes in common with envelope protein gp120 of human immunodeficiency virus type 1 (HIV-1) and can be detected either by an epitope-specific anti-HIV-1 gp120 monoclonal antibody (MAb) (25–27), or by MAb RAK-BrI, which is directed against breast cancer (28, 29). RAK antigens are absent in normal breast tissue and other normal tissues (28), which suggests the strong diagnostic potential of these unique markers. Very recently, RAK markers were also found in prostate cancer and in a number of benign hyperplasia cases (31).
One of the RAK antigens, p160, corresponding in size to HIV-1 gp160 (precursor of gp120 and gp41), was detected in the blood of 70% of gynecological cancer patients before surgery, in 40 to 50% of breast cancer patients after surgery, in 20% of healthy women with a family history of breast cancer, and in 13% of healthy women without a family history of breast or gynecological cancer (29, 30).
The similarity of RAK breast cancer antigens to HIV-1 major proteins led to speculation that HIV-1-like DNA sequences encoding RAK antigens might also be localized in cancer DNA. That hypothesis was confirmed by the finding that HIV-1 gp41-derived primers SK68 and SK69 initiated a PCR with breast cancer DNA but not with normal breast DNA (28). It is noteworthy that the same HIV-1-derived primers amplified prostate cancer DNA but not normal prostate DNA (31). The prostate cancer DNA fragments amplified exhibited strong homology to HIV-1 (31). The study described in this report revealed strong homology between HIV-1 and breast cancer DNA sequences. Viral particles cross-reactive with the anti-HIV-1 gp120 MAb were also detected in breast cancer cells, supporting the hypothesis of a potential viral origin of breast cancer.
I don’t see any #BlotGate type shenanigans here:
FIG. 2 Reactivity of an anti-HIV-1 gp120 MAb with breast and gynecological cancer cytoplasmic proteins and HIV-1 proteins. (A) Lanes: 1 and 2, normal breast and normal uterine proteins, respectively; 3 to 5, breast cancer tissues from three different patients; 6, uterine cancer; 7 and 8, HIV-1 gp160 and gp120, respectively; 9, HIV-1 p24 (negative control). The anti-HIV-1 MAb reacted with three cancer proteins (RAK p120, p42, and p25) but also with HIV-1 gp120 and its precursor gp160. The positions of p120, p42, and p24 are shown on the left. (B) Reactivity of the anti-HIV-1 gp120 MAb with breast cancer proteins obtained from two different patients (lanes 1 and 2) before and after preincubation with the indicated peptides. The peptides containing the consensus sequence GRAF or GRVV inhibited the binding of the anti-HIV-1 MAb to all three cancer antigens. Positively charged lysine in the peptide GRKF did not allow MAb binding, and the peptide did not affect interaction with cancer antigens.
Electron microscopic detection of viral particles.
Electron microscopic analysis of thin sections of SiHa cervical cancer and MCF 7 breast cancer cells revealed large, membrane-coated vesicles (vacuoles) that were immunogold labeled with the anti-HIV-1 gp120 MAb and localized in the cytoplasm (Fig. (Fig.5A),5A), at the edges of cells (Fig. (Fig.5B),5B), and outside of cells (Fig. (Fig.5C).5C). Strong immunogold labeling of the villus-like structures on the cell surface, as well as of the intracellular and extracellular particles, was also observed (Fig. (Fig.5A).5A).
FIG.5 Transmission electron micrographs of cellular and extracellular vacuoles carrying viral particles in SiHa (A, C, and D) and MCF 7 (B) cells. Viral particles were obtained by ultracentrifugation (100,000 × g, 1 h) of cell culture media and negatively stained with uranyl acetate (E to G). In A and D to G, samples were also immunogold labeled with an anti-HIV-1 gp120 MAb. The sizes of the immunogold particles (arrows in A) were 15 (A and D) and 10 (E) nm. V, virus-like particles. Bars, 100 nm. The original magnification was 30,000 (C) or 75,000 (A, B, and D to G).
DISCUSSION
RAK antigens p120, p42, and p25 are expressed in breast and gynecological cancer tissues in women (28, 29) and prostate cancer tissue in men (31) but are absent in normal tissues and in the majority of NATs. Although the nature of these unique proteins is not fully understood, the unusual association with cancer strongly suggests a potential role in the etiology of cancers of the reproductive system. Why cancers that differ in histological structure and originated from completely different tissues all express these unique proteins remains a medical and biological puzzle. If we assume that RAK antigens are encoded by a virus, then one of the possible mechanisms is hormonal regulation. One of the cancer antigens (RAK antigen p160) is expressed in the blood of the majority of breast, cervical, and ovarian cancer patients (29, 30). The molecular weight correlation of blood RAK antigen p160 with the precursor of HIV-1 envelope proteins gp120 and gp41 strongly supports a viral origin of this marker. Cancer-limited expression of RAK antigens suggests either that normal human genes are selectively transcribed in cancer or that a unique virus affects reproductive organs, leading to malignancy. Transcriptional regulation of human genes is very unlikely in light of the fact that HIV-1 gp41-derived primers PCR amplified breast (28) and prostate (31) cancer DNAs but not normal tissue DNA, including DNA extracted from NAT.
In 48% of breast cancer patients, HIV-1-like sequences were absent in histologically normal tissue of the cancer-affected breast, which eliminates the possibility of a random distribution of these sequences in the human genome and excludes the possibility that the identified sequences were of human origin. Whether the identified HIV-1 gp41-like sequences encode RAK antigen p42 cannot be established, but it seems very unlikely that HIV-1-like RAK antigens and HIV-1-like DNA sequences represent two unrelated phenomena, both exclusively associated with cancer.
Breast cancer RAK antigens p120, p42, and p25 exhibit molecular and immunological similarity to the proteins encoded by HIV-1. Moreover, RAK antigens express an amino acid region homologous to variable loop V3 of HIV-1 and cross-react with an epitope-specific anti-HIV-1 envelope protein gp120 MAb. MAb RAK-BrI, which was developed against RAK antigens, also cross-reacts with HIV-1 antigens gp160 and gp120, which confirms the nonaccidental similarity of cancer and HIV-1 proteins. Recent studies indicated that several antibodies developed against a nonglycosylated form of HIV-1 gp120 recognized RAK p120 in cancer cells, which implies that the homology of RAK antigen p120 to HIV-1 gp120 is expanded to various parts of the molecule but is probably limited to the primary structure of the protein (31a). Previous studies indicated that a MAb raised against HIV-1 gp41 is also able to recognize RAK p42 in breast and cervical cancer cell lines; however, limited studies were done with that MAb (27).
Although RAK antigens exhibit homology to HIV-1 antigens, these cancer markers can be easily distinguished from HIV-1 infection by using (i) any antibody directed against the glycosylated form of HIV-1 gp120, (ii) MAb 5025, or (iii) any other anti-HIV-1 MAb which does not cross-react with cancer antigens. Moreover, MAb RAK-BrI binds to RAK antigens p120, p42, and p25 in cancer tissue but only to gp160 and gp120 of HIV-1, which automatically eliminates the possibility of infection.
The similarity of breast cancer RAK antigens p120, p42, and p25 to HIV-1 major proteins, the fact that all three antigens are usually found together, and the exclusive cancer affiliation of these markers, further supported by the presence of HIV-1-like sequences in cancer DNA, strongly suggest that these antigens belong to a slow retrovirus, a fragment of which has been sequenced. Inhibition of cancer cell growth, in parallel with inhibition of reverse transcriptase activity in the presence of antisense oligonucleotides complementary to the HIV-1-like sequences, supports the viral nature of RAK markers. The mechanism of cancer growth promotion by the putative virus remains to be further investigated. The growth factor-like character of the RAK antigens is suggested by the previously described cancer growth activation of the anti-HIV-1 gp120 MAb (27).
The presence of HIV in cell cultures or in fresh cancer tissue may be eliminated since they tested negative for HIV-1 p24.The fact that of the over 1,000 cancer patients tested in this and other studies (28, 29) for the presence of RAK antigens in breast and gynecological tissue, 95% were RAK positive automatically excludes the HIV-1 origin of RAK antigens. Further studies are needed to fully characterize and classify the virus.
Independently of the viral or human origin of the novel cancer antigens, the specific association of RAK antigens with gynecological and breast cancer in women and prostate cancer in men and the lack of these markers in normal tissues strongly suggest that RAK markers have a critical value in the early diagnosis of cancers affecting reproductive organs. The diagnostic value of protein and PCR RAK markers was evaluated before (28, 31). It is suggested that protein RAK markers might improve the early detection of malignant or premalignant changes and would help in more effective evaluation of cancer margins, leading to a reduction in the number of unneeded mastectomies. PCR markers could be used to determine the predisposition of breast tissue to become malignant. Current studies on the identification of HIV-1-like sequences will definitely help to clone the virus and understand the etiology of reproductive tract cancers. If the structure of the new cancer virus were understood, then completely new approaches to cancer diagnosis, prevention, prognosis, and therapy could be developed. Cancer RAK antigen p120 and/or other RAK antigens would definitely play a critical role in the production of a breast cancer vaccine.
ACKNOWLEDGMENTS
This study was sponsored by the Leland J. and Dorothy H. Olson Foundation for Women’s Health.
As a fact check I did a summary web search and couldn’t find any calls for retraction or generally asking what the heck?
21 years after this study Polansky & Schwab published a review into how latent viruses may cause breast cancer, lending additional credence to the “cancer virus” hypothesis and experimental findings.
They refer to viruses including Epstein–Barr (EBV), cytomegalovirus (CMV), herpes simplex virus 1 (HSV-1), human immunodeficiency virus (HIV) and human T-cell lymphotropic virus (HTLV). Incidentally they don’t cite studies into RAK antigens - if you don’t look, you won’t find.
They hypothesise that latent viruses may contribute to cancer promotion and progression by competing for a transcription factor called GABP that transactivates the BRCA1 gene, in addition to other pro-cancer pathways.
Pluta et al (2020) go further and reference HIV binding to GABP:
…HIV-1 variant CRF01_AE included a single copy of NF-κB binding site, since the second NF-κB site, usually observed in other subtypes, was replaced by GA-binding protein (GABP) binding site [92,93]37
“How latent viruses cause breast cancer: An explanation based on the microcompetition model” (2019):38
Abstract
Most breast cancer cases show a decrease in the concentration of the breast cancer type 1 susceptibility protein (BRCA1). However, only a small portion of these cases have a mutated BRCA1 gene. Although many attempts have been made to identify the reason for the decrease in BRCA1 concentration in sporadic, non-heritable breast cancer cases, the cause is still unknown. In this review, we use the Microcompetition Model to explain how certain latent viruses, which are frequently detected in breast cancer tumors, can decrease the expression of the BRCA1 gene and cause the development of breast tumors.
In this paper, we use the Microcompetition Model to show how certain latent viruses, which are frequently detected in breast cancer, can decrease the expression of the BRCA1 gene and cause the development of breast tumors.
They then discussed how most cases of breast cancer aren’t caused by heritable mutations of BRCA1/2 or hypermethylation. This leaves oncogenic viruses as the probable causative agents for many incidents:
MMTV: mouse mammary tumour virus.
In case you were wondering, the answer is probably “yes”:
The mouse mammary tumor virus (MMTV) has formerly been classified as a simple retrovirus; however, it has recently been established, that MMTV encodes an extra self-regulatory mRNA export protein, Rem, with resemblance to the human immunodeficiency virus (HIV) Rev protein, and is therefore the first complex murine retrovirus to be documented39.
One study screened DNA samples of 80 Pakistani breast cancer patients for MMTV gene sequences and found that up to 26% of the samples were positive for the presence of the MMTV envelope and long terminal repeat (LTR) sequences [17]. These results indicate a possible association between breast cancer and MMTV.
Other studies found human papillomaviruses (HPV) and the Epstein-Barr virus (EBV) in breast tumors [18-21].
A systematic review and meta-analysis of 29 studies that included 2211 breast tissue samples from across the globe found that 23% of breast cancer patients had HPV DNA compared to 12.9% controls [20]. Also, the researchers pooled the data of nine case-control studies and calculated an odds ratio (OR) of 5.9, indicating that HPV positive women are 5.9 times more likely to have breast cancer [20]. Furthermore, a case-control study in Northern Iran, including 130 individuals, used polymerase chain reaction (PCR) analysis and detected HPV DNA in 25.9% of breast cancer tumors compared to 2.4% in non-cancer breast tissue, where 53% and 0% were the “high risk” HPV subtypes, such as HPV-16 and 18, in breast cancer tumors and non-cancer breast tissue, respectively [21]. The high prevalence of HPV-positive DNA in breast cancer patients suggests a possible link between HPV and breast cancer. In addition, it has been shown that the E6 and E7 oncoproteins of HPV-16 and 18 directly interact with and inactivate BRCA1 in breast cancer cells [22].
Studies also found EBV in breast cancer patients. One European study, which included 196 breast cancer specimens, found EBV DNA in 33.2% of the cases using real-time quantitative PCR (real-time PCR). Interestingly, the EBV-positive breast cancers tended to be tumors with a more aggressive phenotype. These EBV-positive tumors were also more frequently estrogen receptor negative and had a higher histological grade [23]. A large meta-analysis of 24 studies, which included 1535 cases from all over the world, found an EBV infection in 29.3% of the patients with breast cancer. Also, patients with a positive EBV status showed a significant increase in breast malignancy risk (OR = 6.3) [24]. These studies provide evidence that EBV is statistically associated with an increased risk of breast cancer, especially of some specific types of breast cancer, such as lobular breast carcinoma [24].
Viruses have not only been implicated in breast cancer but they have also been linked to several other types of cancers. Most notably, HPV is known to be a necessary factor for the development of cervical cancers [25]. Increasing evidence linking EBV and colorectal cancer (CRC) has emerged. In a study of 90 CRC specimens, EBV proteins were detected in nearly a third of the tissues, compared to a detection rate of only 4% in adjacent non-cancerous control specimens [26]. Another study used PCR and tissue microarray (TMA) analysis to show that EBV was present in 36% of 102 CRC tissue samples, and EBV was also associated with a more aggressive type of CRC [27]. Recent research suggests that a coinfection of EBV and HPV may play an important role in the progression of cervical cancer [28]. The study found that EBV and high-risk HPV were copresent in 34% of the 44 cervical cancer tissues sampled, and the cancers with both infections were likely to be more aggressive [28].
FIGURE 1 The process by which external and internal events lead to breast cancer by way of GABP dysregulation at the BRCA1 promoter. External/internal events cause A) increased latent virus copy number; B) decreased GABP•CBP/p300 binding at BRCA1 promoter; C) decreased BRCA1 gene expression and BRCA1 protein; D) development of breast cancer. GABP: GA-binding protein; BRCA1: Breast cancer type 1 susceptibility protein.
CONCLUSION
The Microcompetition Model shows how an increase in the copy number of the latent virus that infects breast cancer tissues increases the sequestering of the limiting GABP•CBP p300 transcription complex. This disrupts the allocation of the transcription complex to cellular genes, specifically the tumor suppressor BRCA1, which decreases the levels of the BRCA1 protein and causes the development of breast cancer.
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
And we now know from the in silico binding energy studies that gp120 also has a strong binding affinity to BRCA1 itself, if not the precursor GABP.
Ovarian Cancer Latency Period
From the NHS guide “Predictive genetic tests for cancer risk genes”40 the risk of ovarian cancer is also significantly increased by having defective or deficient BRCA proteins. But as per the previously discussed papers other factors are involved in most cases.
About BRCA1 and BRCA2
If you have a fault (mutation) in one of the BRCA genes, your risk of developing breast cancer and ovarian cancer is greatly increased.
Women with the faulty BRCA1 gene, for example, have a 65 to 79% lifetime risk of breast cancer and a 36 to 53% risk of ovarian cancer before the age of 80. In other words, out of every 100 women with the faulty BRCA1 gene, between 65 and 79 will develop breast cancer in their lifetime and between 36 and 53 will develop ovarian cancer.
The faulty BRCA genes affect around 1 in every 300 to 400 people, but people of Ashkenazi Jewish descent are at a much higher risk (as many as 1 in 40 may carry the faulty gene).
The latency period for ovarian cancer is unknown but estimated at 15-20 years41.
Further Support
Cancer support or advice is available from the prostate community trials groups. But please be aware these are staffed only by volunteers, demand is extremely high and additional medical and other support is always welcome:
Knowledge is power, the sooner you can change your lifestyle or introduce more therapeutics into your regime the better.
And advice to everyone, as we have all been exposed to spike protein or oncogenic viruses etc at some point, please get yourself checked out at the earliest opportunity if you experience any unusual signs or symptoms.
DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. Without your support I would be unable maintain this Substack. To receive new posts and support my work, consider becoming a free or paid subscriber.
Big Country - Post Nuclear Talking Blues.
Pun intended.
Disclaimer
This site is strictly an information website about pathologies, potential preventative therapeutics and a narrative review of the current state of research. It does not advertise anything, provide specific medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
Jiang H, Mei YF. SARS-CoV-2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro. Viruses. 2021;13(10):2056. doi:10.3390/v13102056
Sattar S, Kabat J, Jerome K, Feldmann F, Bailey K, Mehedi M. Nuclear translocation of spike mRNA and protein is a novel feature of SARS-CoV-2. Frontiers in Microbiology. 2023;14. Accessed June 10, 2023. https://www.frontiersin.org/articles/10.3389/fmicb.2023.1073789
Ravi R, Piva TJ, Ravi R, Piva TJ. The Role of Furin in the Development of Skin Cancer. In: Highlights in Skin Cancer. IntechOpen; 2013. doi:10.5772/55569
Pradhan P, Pandey A, Mishra A, et al. Uncanny Similarity of Unique Inserts in the 2019-NCoV Spike Protein to HIV-1 Gp120 and Gag.; 2020. doi:10.1101/2020.01.30.927871
Tamamis P, Floudas CA. Molecular Recognition of CXCR4 by a Dual Tropic HIV-1 gp120 V3 Loop. Biophys J. 2013;105(6):1502-1514. doi:10.1016/j.bpj.2013.07.049
López SN, Rodríguez-Valentín M, Rivera M, et al. HIV-1 Gp120 clade B/C induces a GRP78 driven cytoprotective mechanism in astrocytoma. Oncotarget. 2017;8(40):68415-68438. doi:10.18632/oncotarget.19474
Berth S, Caicedo HH, Sarma T, Morfini G, Brady ST. Internalization and Axonal Transport of the HIV Glycoprotein gp120. ASN Neuro. 2015;7(1):1759091414568186. doi:10.1177/1759091414568186
Berth SH, Mesnard-Hoaglin N, Wang B, et al. HIV Glycoprotein Gp120 Impairs Fast Axonal Transport by Activating Tak1 Signaling Pathways. ASN Neuro. 2016;8(6):1759091416679073. doi:10.1177/1759091416679073
Masimani V, Ravindrana Y, Venugopal G, Raghavendran S, Selvaraj S, Chandrasekaran D. SARS-CoV-2 Kerala Isolate Spike Protein Induces Cancer Proliferating Markers for Lung and Breast Cancer: An In Silico Approach. Coronaviruses. 2022;3(4):69-80. doi:10.2174/2666796703666220518152248
Begum F, Mukherjee D, Thagriki D, et al. Analyses of spike protein from first deposited sequences of SARS-CoV2 from West Bengal, India. F1000Research. 2020;9:371. doi:10.12688/f1000research.23805.1
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server issue):W363-367. doi:10.1093/nar/gki481
Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: a web server for fast interaction refinement in molecular docking†. Nucleic Acids Res. 2008;36(Web Server issue):W229-W232. doi:10.1093/nar/gkn186
Turner JS, O’Halloran JA, Kalaidina E, et al. SARS-CoV-2 mRNA vaccines induce persistent human germinal centre responses. Nature. 2021;596(7870):109-113. doi:10.1038/s41586-021-03738-2
Patters BJ, Kumar S. The role of exosomal transport of viral agents in persistent HIV pathogenesis. Retrovirology. 2018;15:79. doi:10.1186/s12977-018-0462-x
Ivanov AV, Valuev-Elliston VT, Tyurina DA, et al. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis.Oncotarget. 2016;8(3):3895-3932. doi:10.18632/oncotarget.13904
Isaguliants M, Bayurova E, Avdoshina D, Kondrashova A, Chiodi F, Palefsky JM. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers (Basel). 2021;13(2):305. doi:10.3390/cancers13020305
Saftig P, Hetman M, Schmahl W, et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14(15):3599-3608.
Lasky T, Silbergeld E. P53 mutations associated with breast, colorectal, liver, lung, and ovarian cancers.Environ Health Perspect. 1996;104(12):1324-1331. doi:10.1289/ehp.961041324
Liu Z, Zang Y, Qiao L, et al. ASPP2 involvement in p53-mediated HIV-1 envelope glycoprotein gp120 neurotoxicity in mice cerebrocortical neurons. Sci Rep. 2016;6:33378. doi:10.1038/srep33378
Al-Yousef N, Shinwari Z, Al-Shahrani B, Al-Showimi M, Al-Moghrabi N. Curcumin induces re-expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines. Oncol Rep. 2020;43(3):827-838. doi:10.3892/or.2020.7473
Rakowicz-Szulczynska EM, Jackson B, Szulczynska AM, Smith M. Human Immunodeficiency Virus Type 1-Like DNA Sequences and Immunoreactive Viral Particles with Unique Association with Breast Cancer.Clin Diagn Lab Immunol. 1998;5(5):645-653.
Pluta A, Jaworski JP, Cortés-Rubio CN. Balance between Retroviral Latency and Transcription: Based on HIV Model. Pathogens. 2021;10(1):16. doi:10.3390/pathogens10010016
Polansky H, Schwab H. How latent viruses cause breast cancer: An explanation based on the microcompetition model. Bosn J Basic Med Sci. 2019;19(3):221-226. doi:10.17305/bjbms.2018.3950
Tokuoka S, Kawai K, Shimizu Y, et al. Malignant and benign ovarian neoplasms among atomic bomb survivors, Hiroshima and Nagasaki, 1950-80.J Natl Cancer Inst. 1987;79(1):47-57.
Excellent review thanks. We are gradually undertanding the mechanims involved in AEs from the spikes, which are many and complex. I personally find the work on IgG3/4 also compelling. We remain limited in our total understanding by virtue of the fact that we still probably understand only about 10% of the human immune system.






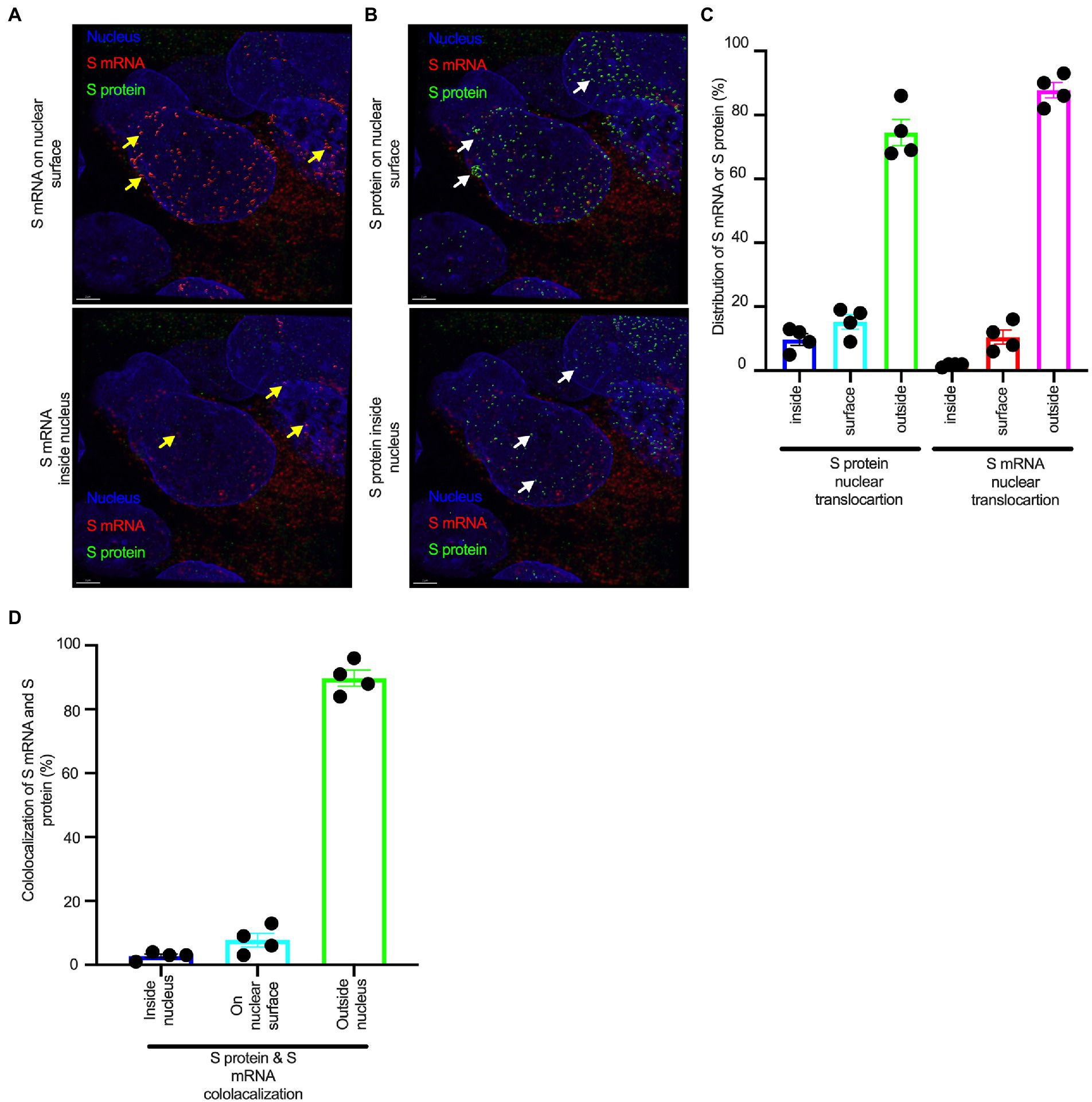

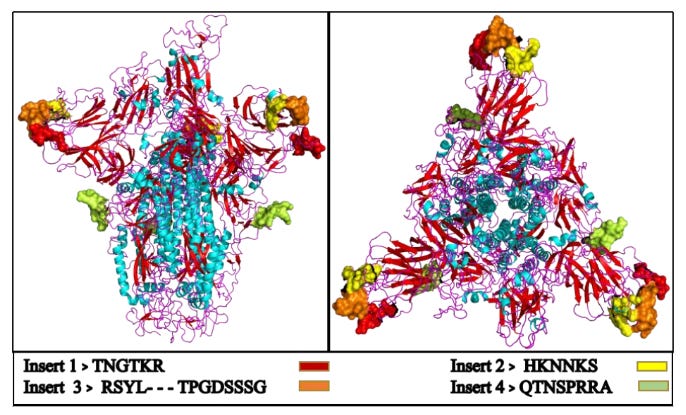


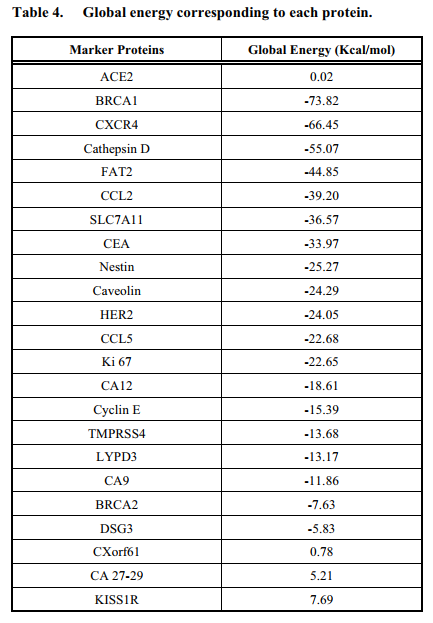
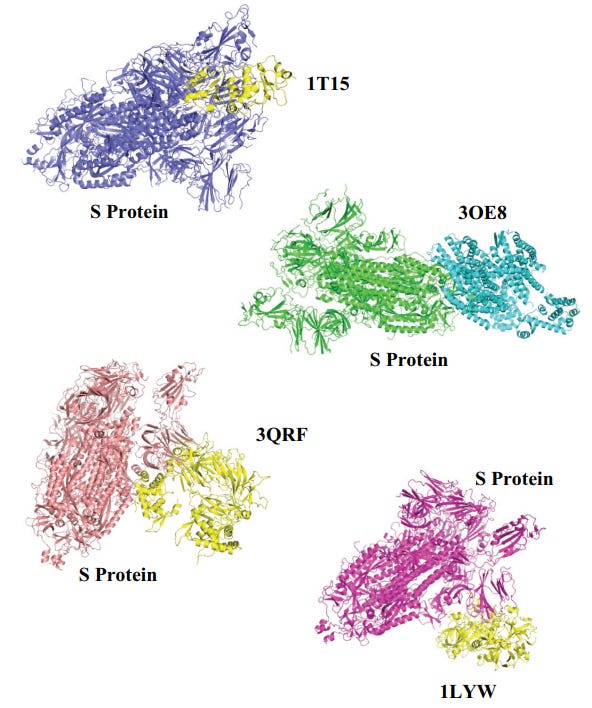













Excellent review thanks. We are gradually undertanding the mechanims involved in AEs from the spikes, which are many and complex. I personally find the work on IgG3/4 also compelling. We remain limited in our total understanding by virtue of the fact that we still probably understand only about 10% of the human immune system.
No medical school goes into such depth , nor do oncologists or radiation oncologists know this . Thank you for the education.