This substack is based on the knowledge base I established for A Prostate Community Trial. It needs to be shared as widely as possible - a public substack is one the best ways to do this both for the trials group going forward and a wider audience. Certain case related details have been removed to maintain confidentiality.
This is by no means a comprehensive list as many other therapeutics also have significant anticancer or preventative properties and may possibly be combined to gain synergistic benefits, subject to interactions checks and advice from a qualified medical adviser.
Document updates will appear here as soon as I have completed their reviews, please keep checking back.
If you or anyone you know would like further support please read the following and click on the link:
This site is strictly an information website about potential therapeutic agents and a review of the current state of research. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
Useful references collated in “the Foolishness of God” website: Ivermectin vs. Cancer
Some of these will be duplicates, none of them have abstracts or a synopsis – I will add these later but feel it important to get them recorded in case this excellent site gets taken down. Many thanks must go to the author for collating all these:
Monday, September 5, 2022
This page grew out of my Ivermectin vs. COVID-19 page. While collecting links on that subject I ran across these papers on ivermectin and cancer.
The drug shows some amazing potential as a safe, non-toxic, treatment that can be used as an alternative, or addition, to chemotherapy.
Antitumor effects of ivermectin at clinically feasible concentrations support its clinical development as a repositioned cancer drug. (2020)
Synopsis:
An in vitro study of ivermectin against 28 malignant cell lines, and an in vivo study of the antitumor effects using a mouse model.
Abstract
Purpose: Ivermectin is an antiparasitic drug that exhibits antitumor effects in preclinical studies, and as such is currently being repositioned for cancer treatment. However, divergences exist regarding its employed doses in preclinical works. Therefore, the aim of this study was to determine whether the antitumor effects of ivermectin are observable at clinically feasible drug concentrations.
Methods: Twenty-eight malignant cell lines were treated with 5 μM ivermectin. Cell viability, clonogenicity, cell cycle, cell death and pharmacological interaction with common cytotoxic drugs were assessed, as well as the consequences of its use on stem cell-enriched populations. The antitumor in vivo effects of ivermectin were also evaluated.
Results: The breast MDA-MB-231, MDA-MB-468, and MCF-7, and the ovarian SKOV-3, were the most sensitive cancer cell lines to ivermectin. Conversely, the prostate cancer cell line DU145 was the most resistant to its use. In the most sensitive cells, ivermectin induced cell cycle arrest at G0-G1 phase, with modulation of proteins associated with cell cycle control. Furthermore, ivermectin was synergistic with docetaxel, cyclophosphamide and tamoxifen. Ivermectin reduced both cell viability and colony formation capacity in the stem cell-enriched population as compared with the parental one. Finally, in tumor-bearing mice ivermectin successfully reduced both tumor size and weight.
Conclusion: Our results on the antitumor effects of ivermectin support its clinical testing.
Keywords: Cancer; Cancer stem cells; Drug repurposing; Ivermectin.
Juarez M, Schcolnik-Cabrera A, Dominguez-Gomez G, Chavez-Blanco A, Diaz-Chavez J, Duenas-Gonzalez A. Antitumor effects of ivermectin at clinically feasible concentrations support its clinical development as a repositioned cancer drug. Cancer Chemother Pharmacol. 2020 Jun;85(6):1153-1163. doi: 10.1007/s00280-020-04041-z. Epub 2020 May 30. PMID: 32474842.
Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. (2020)
Synopsis
Phosphorylation activates HSP27, a heat shock protein, that then can activate an oncogene that stops a cell from dying via apoptosis, a key step in becoming cancerous.
Heat shock proteins (HSP) are a family of proteins that are produced by cells in response to exposure to stressful conditions. They are emerging as important molecules in the development of cancer and as key targets in cancer therapy. These proteins enhance the growth of cancer cells and protect tumors from treatments such as drugs or surgery, otherwise known as multidrug resistance.
Epithelial-mesenchymal transition (EMT)is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells.
“IVM is a member of the avermectin family of macrocyclic lactones (herein called IVM analogs) that similarly inhibit HSP27 in FRET and FP assays.”
In silico and in vivo studies involving mice appeared to confirm the hypothesis that IVM inhibits HSP27 phosphorylation and EMT. Other anticancer properties were also described in the discussion.
Abstract
HSP27 is highly expressed in, and supports oncogene addiction of, many cancers. HSP27 phosphorylation is a limiting step for activation of this protein and a target for inhibition, but its highly disordered structure challenges rational structure-guided drug discovery. We performed multistep biochemical, structural, and computational experiments to define a spherical 24-monomer complex composed of 12 HSP27 dimers with a phosphorylation pocket flanked by serine residues between their N-terminal domains. Ivermectin directly binds this pocket to inhibit MAPKAP2-mediated HSP27 phosphorylation and depolymerization, thereby blocking HSP27-regulated survival signaling and client-oncoprotein interactions. Ivermectin potentiated activity of anti–androgen receptor and anti-EGFR drugs in prostate and EGFR/HER2-driven tumor models, respectively, identifying a repurposing approach for cotargeting stress-adaptive responses to overcome resistance to inhibitors of oncogenic pathway signaling.
Keywords: Oncology
Keywords: Drug therapy
Nappi L, Aguda AH, Nakouzi NA, Lelj-Garolla B, Beraldi E, Lallous N, Thi M, Moore S, Fazli L, Battsogt D, Stief S, Ban F, Nguyen NT, Saxena N, Dueva E, Zhang F, Yamazaki T, Zoubeidi A, Cherkasov A, Brayer GD, Gleave M. Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. J Clin Invest. 2020 Feb 3;130(2):699-714. doi: 10.1172/JCI130819. PMID: 31845908; PMCID: PMC6994194.
The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. (2010)
Synopsis
This study used a chemical screen to identify drugs with previously unknown activity against leukaemia, and ivermectin showed efficacy. They then went on to conduct in vivo experiments for effective dose and cytotoxicity assessments.
Efficacy against leukaemia and prostate cancer cell lines were assessed in vitro.
Ivermectin inhibits cancer cells using multiple mechanisms, including reactive oxygen species generation (ROS free radicals), an increase in intracellular chloride and by synergy with other allopathic chemotherapy drugs: cytarabine and daunorubicin.
“Thus, given its prior safety and toxicity testing, ivermectin could be rapidly advanced into clinical trial for patients with leukemia.”
Abstract
To identify known drugs with previously unrecognized anticancer activity, we compiled and screened a library of such compounds to identify agents cytotoxic to leukemia cells. From these screens, we identified ivermectin, a derivative of avermectin B1 that is licensed for the treatment of the parasitic infections, strongyloidiasis and onchocerciasis, but is also effective against other worm infestations. As a potential antileukemic agent, ivermectin induced cell death at low micromolar concentrations in acute myeloid leukemia cell lines and primary patient samples preferentially over normal hematopoietic cells. Ivermectin also delayed tumor growth in 3 independent mouse models of leukemia at concentrations that appear pharmacologically achievable. As an antiparasitic, ivermectin binds and activates chloride ion channels in nematodes, so we tested the effects of ivermectin on chloride flux in leukemia cells. Ivermectin increased intracellular chloride ion concentrations and cell size in leukemia cells. Chloride influx was accompanied by plasma membrane hyperpolarization, but did not change mitochondrial membrane potential. Ivermectin also increased reactive oxygen species generation that was functionally important for ivermectin-induced cell death. Finally, ivermectin synergized with cytarabine and daunorubicin that also increase reactive oxygen species production. Thus, given its known toxicology and pharmacology, ivermectin could be rapidly advanced into clinical trial for leukemia.
Sharmeen S, Skrtic M, Sukhai MA, Hurren R, Gronda M, Wang X, Fonseca SB, Sun H, Wood TE, Ward R, Minden MD, Batey RA, Datti A, Wrana J, Kelley SO, Schimmer AD. The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood. 2010 Nov 4;116(18):3593-603. doi: 10.1182/blood-2010-01-262675. Epub 2010 Jul 19. PMID: 20644115.
Ivermectin, a potential anticancer drug derived from an antiparasitic drug. (2020)
Synopsis
If you only read one paper reviewing the multiple mechanisms involved then this is an excellent one to study in full.
This literature review discusses the research into the multiple mechanisms of cancer cell inhibition mediated by ivermectin, explores the chemical structure and related compounds, its role in the inhibition of a wide range of cancer types (breast, gastric, colorectal, hepatocellular, renal, prostate, leukaemia, cervical, ovarian, brain, nasopharynx, lung and melanoma), modes of cell death induction, reversal of multidrug resistance, signalling pathways invoked and effects on tumor stem cells.
Apoptosis: the death of cells which occurs as a normal and controlled part of an organism's growth or development. Also called programmed cell death.
Autophagy: is the natural, conserved degradation of the cell that removes unnecessary or dysfunctional components through a lysosome-dependent regulated mechanism.[3] It allows the orderly degradation and recycling of cellular components.[4][5] Although initially characterized as a primordial degradation pathway induced to protect against starvation, it has become increasingly clear that autophagy also plays a major role in the homeostasis of non-starved cells.[6] Defects in autophagy have been linked to various human diseases, including neurodegeneration and cancer, and interest in modulating autophagy as a potential treatment for these diseases has grown rapidly.
Pyroptosis is a highly inflammatory form of lyticprogrammed cell death that occurs most frequently upon infection with intracellular pathogens and is likely to form part of the antimicrobial response. This process promotes the rapid clearance of various bacterial, viral, fungal and protozoan infections by removing intracellular replication niches and enhancing the host's defensive responses. Pyroptosis can take place in immune cells and is also reported to occur in keratinocytes and some epithelial cells.[1]
Graphical abstract
Ivermectin has powerful antitumor effects, including the inhibition of proliferation, metastasis, and angiogenic activity, in a variety of cancer cells. This may be related to the regulation of multiple signaling pathways by ivermectin through PAK1 kinase. On the other hand, ivermectin promotes programmed cancer cell death, including apoptosis, autophagy and pyroptosis. Ivermectin induces apoptosis and autophagy is mutually regulated. Interestingly, ivermectin can also inhibit tumor stem cells and reverse multidrug resistance and exerts the optimal effect when used in combination with other chemotherapy drugs.
Abstract
Ivermectin is a macrolide antiparasitic drug with a 16-membered ring that is widely used for the treatment of many parasitic diseases such as river blindness, elephantiasis and scabies. Satoshi ōmura and William C. Campbell won the 2015 Nobel Prize in Physiology or Medicine for the discovery of the excellent efficacy of ivermectin against parasitic diseases. Recently, ivermectin has been reported to inhibit the proliferation of several tumor cells by regulating multiple signaling pathways. This suggests that ivermectin may be an anticancer drug with great potential. Here, we reviewed the related mechanisms by which ivermectin inhibited the development of different cancers and promoted programmed cell death and discussed the prospects for the clinical application of ivermectin as an anticancer drug for neoplasm therapy.
Fig.2 Mechanisms of IVM-induced mitochondria-mediated apoptosis. Cancer cells exposure to IVM can be induced to generate ROS generation and reduce membrane potential of mitochondria. Moreover, IVM can up-regulate Bax and down-regulate Bcl-2, promote releasing of cytochrome C into the cytosol, and activate the signaling cascade of caspases–9/3. Finally, activated PARP and caspase-3 trigger apoptosis.
Fig.3 Mechanisms of IVM-induced PAK1/Akt/mTOR-mediated autophagy. IVM promotes degradation of PAK1 by ubiquitination/proteasome pathway, thereby inhibiting the Akt/mTOR signaling pathway. Subsequently, the inactivation Akt/mTOR signaling cannot inhibit the formation of the Beclin-1 complex, thus inducing the formation autophagosome. Overall, IVM can induce autophagy through PAK1/Akt/mTOR pathway to represses the growth of cancer cells independent of apoptosis. (Ub:Ubiquitination, P:Phosphorylation)
Fig.4 Mechanisms of IVM-induced P2 × 4/P2 × 7/NLRP3-mediated pyroptosis. IVM can promote ROS release in cancer cells by P2 × 4/P2 × 7 receptors. Cellular ROS can activate NLRP3 Inflammasome including ASC, NLRP3 and pro-caspase-1 assemble. Subsequently, NLRP3 Inflammasome initiates pro-caspase-1 to self-shear into mature caspase-1. On the one hand, activated caspase-1 induces the secretion of pro-inflammatory cytokines IL-1β and IL-18. On the other hand, caspase-1 activated by GSDMD triggers pyroptosis independent of apoptosis.
Fig.5 PAK1 cross regulate multiple signal pathways. RAS activation directly initiates PAK1, MAPK and PI3K/Akt pathway. PAK1 allocates crosstalk between the PI3K and MAPK pathways. PAK1 can induce MEK1/2 and ERK1/2 activation by RAF and increase PI3K/Akt signaling by PDK1. PAK1 can also activate pro-inflammatory pathways by facilitating nuclear activation of NF-kappa B. In addition, PAK1 facilitates Wnt/β-catenin signaling, make β-catenin accumulate in the cytoplasm and translocate to the nucleus. Moreover, Akt can inhibit β-catenin transfer into nucleus.
6. Summary and outlooks
Malignant tumors are one of the most serious diseases that threaten human health and social development today, and chemotherapy is one of the most important methods for the treatment of malignant tumors. In recent years, many new chemotherapeutic drugs have entered the clinic, but tumor cells are prone to drug resistance and obvious adverse reactions to these drugs. Therefore, the development of new drugs that can overcome resistance, improve anticancer activity, and reduce side effects is an urgent problem to be solved in chemotherapy. Drug repositioning is a shortcut to accelerate the development of anticancer drugs.
As mentioned above, the broad-spectrum antiparasitic drug IVM, which is widely used in the field of parasitic control, has many advantages that suggest that it is worth developing as a potential new anticancer drug. IVM selectively inhibits the proliferation of tumors at a dose that is not toxic to normal cells and can reverse the MDR of tumors. Importantly, IVM is an established drug used for the treatment of parasitic diseases such as river blindness and elephantiasis. It has been widely used in humans for many years, and its various pharmacological properties, including long- and short-term toxicological effects and drug metabolism characteristics are very clear. In healthy volunteers, the dose was increased to 2 mg/Kg, and no serious adverse reactions were found, while tests in animals such as mice, rats, and rabbits found that the median lethal dose (LD50) of IVM was 10-50 mg/Kg [112] In addition, IVM has also been proven to show good permeability in tumor tissues [50]. Unfortunately, there have been no reports of clinical trials of IVM as an anticancer drug. There are still some problems that need to be studied and resolved before IVM is used in the clinic.
(1) Although a large number of research results indicate that IVM affects multiple signaling pathways in tumor cells and inhibits proliferation, IVM may cause antitumor activity in tumor cells through specific targets. However, to date, no exact target for IVM action has been found. (2) IVM regulates the tumor microenvironment, inhibits the activity of tumor stem cells and reduces tumor angiogenesis and tumor metastasis. However, there is no systematic and clear conclusion regarding the related molecular mechanism. Therefore, in future research, it is necessary to continue to explore the specific mechanism of IVM involved in regulating the tumor microenvironment, angiogenesis and EMT. (3) It has become increasingly clear that IVM can induce a mixed cell death mode involving apoptosis, autophagy and pyroptosis depending on the cell conditions and cancer type. Identifying the predominant or most important contributor to cell death in each cancer type and environment will be crucial in determining the effectiveness of IVM-based treatments. (4) IVM can enhance the sensitivity of chemotherapeutic drugs and reduce the production of resistance. Therefore, IVM should be used in combination with other drugs to achieve the best effect, while the specific medication plan used to combine IVM with other drugs remains to be explored.
Most of the anticancer research performed on the avermectin family has been focused on avermectin and IVM until now. Avermectin family drugs such as selamectin [36,41,113], and doramectin [114] also have anticancer effects, as previously reported. With the development of derivatives of the avermectin family that are more efficient and less toxic, relevant research on the anticancer mechanism of the derivatives still has great value. Existing research is sufficient to demonstrate the great potential of IVM and its prospects as a novel promising anticancer drug after additional research. We believe that IVM can be further developed and introduced clinically as part of new cancer treatments in the near future.
Tang M, Hu X, Wang Y, Yao X, Zhang W, Yu C, Cheng F, Li J, Fang Q. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol Res. 2021 Jan;163:105207. doi: 10.1016/j.phrs.2020.105207. Epub 2020 Sep 21. PMID: 32971268; PMCID: PMC7505114.
Integrated analysis reveals FOXA1 and Ku70/Ku80 as targets of ivermectin in prostate cancer. (2022)
Synopsis
Further mechanisms of cancer cell death induction by ivermectin are by its interaction with the forkhead box protein and by preventing DNA double-strand break repairs, thus preventing cell division and instead causing it to die.
G0/G1 cell cycle arrest: “G0 to G1 Phase Arrest consists of interference with, or restraint of, activities that regulate cellular capability to transition from the resting stage (between successive cell divisions) and the initial phase of the cell cycle (Gap 1 phase), preceding DNA synthesis. G1 subphases include competence, entry (G1a), progression (G1b), and assembly (G1c), based on effects of limiting growth factors, nutrients, or inhibitors.”
Forkhead box protein A1: “Forkhead box protein A1 (FOXA1) is a transcription factor; recent studies have reported that FOXA1 has an oncogenic or tumor suppressive role in human malignancies, and its expression is associated with the prognosis of patients with cancer.”
Ku70/Ku80: “Ku70 is a DNA repair subunit protein that binds to DNA double-strand break ends and helps repair DNA via the non-homologous end-joining (NHEJ) pathway.”
Abstract
Ivermectin is a widely used antiparasitic drug and shows promising anticancer activity in various cancer types. Although multiple signaling pathways modulated by ivermectin have been identified in tumor cells, few studies have focused on the exact target of ivermectin. Herein, we report the pharmacological effects and targets of ivermectin in prostate cancer. Ivermectin caused G0/G1 cell cycle arrest, induced cell apoptosis and DNA damage, and decreased androgen receptor (AR) signaling in prostate cancer cells. Further in vivo analysis showed ivermectin could suppress 22RV1 xenograft progression. Using integrated omics profiling, including RNA-seq and thermal proteome profiling, the forkhead box protein A1 (FOXA1) and non-homologous end joining (NHEJ) repair executer Ku70/Ku80 were strongly suggested as direct targets of ivermectin in prostate cancer. The interaction of ivermectin and FOXA1 reduced the chromatin accessibility of AR signaling and the G0/G1 cell cycle regulator E2F1, leading to cell proliferation inhibition. The interaction of ivermectin and Ku70/Ku80 impaired the NHEJ repair ability. Cooperating with the downregulation of homologous recombination repair ability after AR signaling inhibition, ivermectin increased intracellular DNA double-strand breaks and finally triggered cell death. Our findings demonstrate the anticancer effect of ivermectin in prostate cancer, indicating that its use may be a new therapeutic approach for prostate cancer.
Lv S, Wu Z, Luo M, Zhang Y, Zhang J, Pascal LE, Wang Z, Wei Q. Integrated analysis reveals FOXA1 and Ku70/Ku80 as targets of ivermectin in prostate cancer. Cell Death Dis. 2022 Sep 1;13(9):754. doi: 10.1038/s41419-022-05182-0. PMID: 36050295; PMCID: PMC9436997.
Anti-Androgen Therapy Can Fuel Spread of Bone Tumors in Prostate Cancer. (2021)
““We wanted to see if the therapy could be a contributor of cancer cells' adaptive responses that fueled bone metastasis,” noted Dietmar Hutmacher, PhD, a Distinguished Professor at Queensland University of Technology and Bock's mentor. “We developed an all-human, microtissue-engineered model of metastatic tissue using human bone-forming cells, prostate cancer cells, and 3-D printing.”
Judith Clements, PhD, a cancer biologist and Distinguished Professor at Queensland University of Technology, said the team bioengineered the microenvironment of a bone tumor to assess the effects of two clinically routinely used anti-androgen therapies—enzalutamide and bicalutamide—on the tumor cells.
“We found that the interactions between the cancer cells, the bone, and the anti-androgens significantly impacted the progress of cancer in the mineralized microenvironment of bone tumors,” Clements stated. “This means that the efficacy of these therapies is compromised in the presence of the bone microenvironment.”
This paper was particularly useful for the diagnostics team for one of our participants.
TNBC is particularly challenging to treat effectively, but ivermectin and lactoferrin can both inhibit EMT (ie metastasis) and reduce multidrug resistance to chemotherapy.
Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer (2015)
Abstract
Triple-negative breast cancers (TNBC) lacking estrogen, progesterone, and HER2 receptors account for 10% to 20% of breast cancer and are indicative of poor prognosis. The development of effective treatment strategies therefore represents a pressing unmet clinical need. We previously identified a molecularly targeted approach to target aberrant epigenetics of TNBC using a peptide corresponding to the SIN3 interaction domain (SID) of MAD. SID peptide selectively blocked binding of SID-containing proteins to the paired α-helix (PAH2) domain of SIN3, resulting in epigenetic and transcriptional modulation of genes associated with epithelial-mesenchymal transition (EMT). To find small molecule inhibitor (SMI) mimetics of SID peptide, we performed an in silico screen for PAH2 domain-binding compounds. This led to the identification of the avermectin macrocyclic lactone derivatives selamectin and ivermectin (Mectizan) as candidate compounds. Both selamectin and ivermectin phenocopied the effects of SID peptide to block SIN3-PAH2 interaction with MAD, induce expression of CDH1 and ESR1, and restore tamoxifen sensitivity in MDA-MB-231 human and MMTV-Myc mouse TNBC cells in vitro. Treatment with selamectin or ivermectin led to transcriptional modulation of genes associated with EMT and maintenance of a cancer stem cell phenotype in TNBC cells. This resulted in impairment of clonogenic self-renewal in vitro and inhibition of tumor growth and metastasis in vivo. Underlining the potential of avermectins in TNBC, pathway analysis revealed that selamectin also modulated the expression of therapeutically targetable genes. Consistent with this, an unbiased drug screen in TNBC cells identified selamectin-induced sensitization to a number of drugs, including those targeting modulated genes.
Kwon YJ, Petrie K, Leibovitch BA, Zeng L, Mezei M, Howell L, Gil V, Christova R, Bansal N, Yang S, Sharma R, Ariztia EV, Frankum J, Brough R, Sbirkov Y, Ashworth A, Lord CJ, Zelent A, Farias E, Zhou MM, Waxman S. Selective Inhibition of SIN3 Corepressor with Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol Cancer Ther. 2015 Aug;14(8):1824-36. doi: 10.1158/1535-7163.MCT-14-0980-T. Epub 2015 Jun 15. PMID: 26078298; PMCID: PMC4529816.
Heat Shock Protein as Molecular Targets for Breast Cancer Therapeutics. (2011)
Triple negative breast cancer (TNBC; defined by the lack of expression of estrogen, progesterone, and HER2) patients have poor prognosis and survival outcomes, but there are currently no specific targeted therapies. Clinical studies have been shown the EGFR overexpression and activation of PI3K pathway in TNBCs and it has been associated with poor prognosis. Hence, HSP90 inhibitors may provide an opportunity to inhibit tumor progression of TNBCs because the many of HSP90 client proteins are oncoproteins including EGFR and involved in multiple oncogenic signaling pathways. Interestingly, PU-H71 (purine based synthetic HSP90 inhibitor) induces tumor regression in a xenograft model of TNBCs and that are not candidate for 17-AAG treatment.
Kim LS, Kim JH. Heat shock protein as molecular targets for breast cancer therapeutics. J Breast Cancer. 2011 Sep;14(3):167-74. doi: 10.4048/jbc.2011.14.3.167. Epub 2011 Sep 29. PMID: 22031796; PMCID: PMC3200510.
EGFR: “A protein found on certain types of cells that binds to a substance called epidermal growth factor. The EGFR protein is involved in cell signaling pathways that control cell division and survival. Sometimes, mutations (changes) in the EGFR gene cause EGFR proteins to be made in higher than normal amounts on some types of cancer cells. This causes cancer cells to divide more rapidly. Drugs that block EGFR proteins are being used in the treatment of some types of cancer. EGFRs are a type of receptor tyrosine kinase. Also called epidermal growth factor receptor, ErbB1, and HER1.”
Our results indicated that ivermectin at its very low dose, which did not induce obvious cytotoxicity, drastically reversed the resistance of the tumor cells to the chemotherapeutic drugs both in vitro and in vivo. Mechanistically, ivermectin reversed the resistance mainly by reducing the expression of P-glycoprotein (P-gp) via inhibiting the epidermal growth factor receptor (EGFR), not by directly inhibiting P-gp activity. Ivermectin bound with the extracellular domain of EGFR, which inhibited the activation of EGFR and its downstream signaling cascade ERK/Akt/NF-κB. The inhibition of the transcriptional factor NF-κB led to the reduced P-gp transcription.
Jiang L, Wang P, Sun YJ, Wu YJ. Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-κB pathway. J Exp Clin Cancer Res. 2019 Jun 18;38(1):265. doi: 10.1186/s13046-019-1251-7. PMID: 31215501; PMCID: PMC6580523.
Use of the Anti-Parasitic Drug Ivermectin to Treat Breast Cancer. (2021)
These promising in vitro results prompted us to move forward to in vivo studies using a common animal model of TNBC. In this model, breast tumors are “cold,” indicating little or no infiltrating T cells. Ivermectin treatment led to robust T-cell infiltration turning cold tumors into hot tumors with cancer cells showing markers of ICD in vivo.
The ability to turn TNBC tumors from cold to hot suggested that ivermectin could synergize with ICI therapy (such as with anti-PD-1 monoclonal antibodies). Immune checkpoint inhibitors block the PD-1 protein, which acts as a brake on T cells, thus helping the immune system do what it is designed to do: eradicate cancer.
Our findings on this novel therapeutic combination published recently in npj Breast Cancer journal (2021; https://doi.org/10.1038/s41523-021-00229-5). This is the first time a research team has demonstrated that checkpoint inhibitors can be used to successfully treat breast cancer—when combined with ivermectin, an inexpensive, existing safe drug.
In these studies, 40-60 percent of animals treated with the ivermectin plus anti-PD1 antibody combination completely eradicated their tumors. They were able to fight off the cancer again after it was reintroduced. It's the two drugs working together that is the magic. Either drug alone has almost zero effect, but together they have a powerful synergistic effect.
Lee, Peter P. MD. Use of the Anti-Parasitic Drug Ivermectin to Treat Breast Cancer. Oncology Times: May 5, 2021 - Volume 43 - Issue 9 - p 10 doi: 10.1097/01.COT.0000751988.88253.c3
Ivermectin, a potential anticancer drug derived from an antiparasitic drug. (2020)
In Nappi's experiment, it was found that IVM could enhance the drug activity of the anti-androgen drug enzalutamide in the prostate cancer cell line LNCaP and reverse the resistance of the prostate cancer cell line PC3 to docetaxel.
Tang M, Hu X, Wang Y, Yao X, Zhang W, Yu C, Cheng F, Li J, Fang Q. Ivermectin, a potential anticancer drug derived from an antiparasitic drug. Pharmacol Res. 2021 Jan;163:105207. doi: 10.1016/j.phrs.2020.105207. Epub 2020 Sep 21. PMID: 32971268; PMCID: PMC7505114.
Other therapeutics to be used individually or for compounding
Lactoferrin
Iron Metabolism in Prostate Cancer; From Basic Science to New Therapeutic Strategies (2018)
An increasing amount of research has recently strengthened the case for the existence of iron dysmetabolism in prostate cancer. It is characterized with a wide array of differential expression of iron-related proteins compared to normal cells. These proteins control iron entry, cellular iron distribution but also iron exit from prostate cells. Iron dysmetabolism is not an exclusive feature of prostate cancer cells, but it is observed in other cells of the tumor microenvironment. Disrupting the machinery that secures iron for prostate cancer cells can retard tumor growth and its invasive potential. This review unveils the current understanding of the ways that prostate cancer cells secure iron in the tumor milieu and how can we exploit this knowledge for therapeutic purposes.
The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators (2021)
Highlights
• Free unbound iron possibly contributes to the hypercoagulation and inflammation found in severe COVID-19.
• The nonapoptotic and immunogenic cell death “ferroptosis” may be a potential contributor to the pathogenesis of COVID-19.
• The bioactive compound lactoferrin and other iron chelators may provide a high therapeutic value in the treatment of COVID-19.
• The relatively lower risk for COVID-19 found in individuals with blood group O may be linked to a lower serum iron status in these individuals.
Abstract
Iron overload is increasingly implicated as a contributor to the pathogenesis of COVID-19. Indeed, several of the manifestations of COVID-19, such as inflammation, hypercoagulation, hyperferritinemia, and immune dysfunction are also reminiscent of iron overload. Although iron is essential for all living cells, free unbound iron, resulting from iron dysregulation and overload, is very reactive and potentially toxic due to its role in the generation of reactive oxygen species (ROS). ROS react with and damage cellular lipids, nucleic acids, and proteins, with consequent activation of either acute or chronic inflammatory processes implicated in multiple clinical conditions. Moreover, iron-catalyzed lipid damage exerts a direct causative effect on the newly discovered nonapoptotic cell death known as ferroptosis. Unlike apoptosis, ferroptosis is immunogenic and not only leads to amplified cell death but also promotes a series of reactions associated with inflammation. Iron chelators are generally safe and are proven to protect patients in clinical conditions characterized by iron overload. There is also an abundance of evidence that iron chelators possess antiviral activities. Furthermore, the naturally occurring iron chelator lactoferrin (Lf) exerts immunomodulatory as well as anti-inflammatory effects and can bind to several receptors used by coronaviruses thereby blocking their entry into host cells. Iron chelators may consequently be of high therapeutic value during the present COVID-19 pandemic.
Hosam M. Habib, Sahar Ibrahim, Aamnah Zaim, Wissam H. Ibrahim, The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators, Biomedicine & Pharmacotherapy, Volume 136, (2021), 111228, ISSN 0753-3322,
Lactoferrin-containing immunocomplex mediates antitumor effects by resetting tumor-associated macrophages to M1 phenotype (2019)
Abstract
Background Tumor-associated macrophages (TAMs) resemble M2-polarized cells with potent immunosuppressive activity and play a pivotal role in tumor growth and progression. Converting TAMs to proinflammatory M1-like phenotype is thus an attractive strategy for antitumor immunotherapy.
Methods A mouse IgG1 (kappa) monoclonal Ab, M-860, specific to human lactoferrin (LTF) was generated by using the traditional hybridoma cell fusion technology. TAMs were generated by culturing human and mouse CD14+ monocytes in tumor-conditioned media containing a cytokine cocktail containing recombinant interleukin-4 (IL-4), interleukin-10 (IL-10) and macrophage colony stimulating factor (M-CSF). TAMs after treatment with immunocomplex (IC) between human LTF and M860 (LTF-IC) were phenotypically and functionally characterized by flow cytometry (FACS), ELISA, Q-PCR and killing assays. The antitumor effects of LTF-IC were further analyzed using in vivo experiments employing tumor-bearing human FcγRIIa-transgenic mouse models.
Results Through coligation of membrane-bound CD14 and FcγRIIa, LTF-IC rendered TAMs not only M2 to M1 conversion, evidenced by increased tumor necrosis factor α production, down-regulated M2-specific markers (CD206, arginase-1 and vascular endothelial growth factor) and upregulated M1-specific markers (CD86 and HLA-DR) expression, but also potent tumoricidal activity in vitro. LTF-IC administration conferred antitumor protective efficacy and prolonged animal survival in FcγRIIa-transgenic mice, accompanied by accumulation of M1-like macrophages as well as significantly reduced infiltration of immunosuppressive myeloid-derived suppressor cells and regulatory T cells in solid tumor tissues.
Conclusions LTF-IC is a promising cancer therapeutic agent capable of converting TAMs into tumoricidal M1-like cells.
Dong H, Yang Y, Gao C, et al, Lactoferrin-containing immunocomplex mediates antitumor effects by resetting tumor-associated macrophages to M1 phenotype, Journal for ImmunoTherapy of Cancer 2020;8:e000339. doi: 10.1136/jitc-2019-000339
Lactoferrin’s Anti-Cancer Properties: Safety, Selectivity, and Wide Range of Action (2020)
Abstract
Despite recent advances in cancer therapy, current treatments, including radiotherapy, chemotherapy, and immunotherapy, although beneficial, present attendant side effects and long-term sequelae, usually more or less affecting quality of life of the patients. Indeed, except for most of the immunotherapeutic agents, the complete lack of selectivity between normal and cancer cells for radio- and chemotherapy can make them potential antagonists of the host anti-cancer self-defense over time. Recently, the use of nutraceuticals as natural compounds corroborating anti-cancer standard therapy is emerging as a promising tool for their relative abundance, bioavailability, safety, low-cost effectiveness, and immuno-compatibility with the host. In this review, we outlined the anti-cancer properties of Lactoferrin (Lf), an iron-binding glycoprotein of the innate immune defense. Lf shows high bioavailability after oral administration, high selectivity toward cancer cells, and a wide range of molecular targets controlling tumor proliferation, survival, migration, invasion, and metastasization. Of note, Lf is able to promote or inhibit cell proliferation and migration depending on whether it acts upon normal or cancerous cells, respectively. Importantly, Lf administration is highly tolerated and does not present significant adverse effects. Moreover, Lf can prevent development or inhibit cancer growth by boosting adaptive immune response. Finally, Lf was recently found to be an ideal carrier for chemotherapeutics, even for the treatment of brain tumors due to its ability to cross the blood–brain barrier, thus globally appearing as a promising tool for cancer prevention and treatment, especially in combination therapies.
Keywords: lactoferrin, cancer, lactoferrin bioavailability, tumor proliferation, apoptosis, epithelial to mesenchymal transition, metastasis, cancer targeting
Cutone A, Rosa L, Ianiro G, Lepanto MS, Bonaccorsi di Patti MC, Valenti P, Musci G. Lactoferrin's Anti-Cancer Properties: Safety, Selectivity, and Wide Range of Action. Biomolecules. 2020 Mar 15;10(3):456. doi: 10.3390/biom10030456. PMID: 32183434; PMCID: PMC7175311.
Lactoferrin: A Glycoprotein Involved in Immunomodulation, Anticancer, and Antimicrobial Processes (2021)
Abstract
Lactoferrin is an iron binding glycoprotein with multiple roles in the body. Its participation in apoptotic processes in cancer cells, its ability to modulate various reactions of the immune system, and its activity against a broad spectrum of pathogenic microorganisms, including respiratory viruses, have made it a protein of broad interest in pharmaceutical and food research and industry. In this review, we have focused on describing the most important functions of lactoferrin and the possible mechanisms of action that lead to its function.
Lactoferrin Exerts Antitumor Effects by Inhibiting Angiogenesis in a HT29 Human Colon Tumor Model (2017)
Abstract
To investigate the effect and potential mechanisms of lactoferrin on colon cancer cells and tumors, HT29 and HCT8 cells were exposed to varying concentrations of lactoferrin, and the impacts on cell proliferation, migration, and invasion were observed. Cell proliferation test showed that high dosage of lactoferrin (5–100 mg/mL) inhibited cell viability in a dose-dependent manner, with the 50% concentration of inhibition at 81.3 ± 16.7 mg/mL and 101 ± 23.8 mg/mL for HT29 and HCT8 cells, respectively. Interestingly, migration and invasion of the cells were inhibited dramatically by 20 mg/mL lactoferrin, consistent with the significant down regulation of VEGFR2, VEGFA, pPI3K, pAkt, and pErk1/2 proteins. HT29 was chosen as the sensitive cell line to construct a tumor-bearing nude mice model. Notably, HT29 tumor weight was greatly reduced in both the lactoferrin group (26.5 ± 6.7 mg) and the lactoferrin/5-Fu group (14.5 ± 5.1 mg), compared with the control one (39.3 ± 6.5 mg), indicating that lactoferrin functioned as a tumor growth inhibitor. Considering lactoferrin also reduced the growth of blood vessels and the degree of malignancy, we concluded that HT29 tumors were effectively suppressed by lactoferrin, which might be achieved by regulation of phosphorylation from various kinases and activation of the VEGFR2-PI3K/Akt-Erk1/2 pathway.
Hui-Ying Li†, Ming Li†, Chao-chao Luo, Jia-Qi Wang*, and Nan Zheng, Food Chem.Lactoferrin Exerts Antitumor Effects by Inhibiting Angiogenesis in a HT29 Human Colon Tumor Mode, J. Agric. (2017), 65, 48, 10464–10472, Publication Date :November 7, 2017
Engineering of Human Lactoferrin for Improved Anticancer Activity (2021)
Abstract
Protease-digested lactoferrin fragments often exhibit improved therapeutic properties. However, there are limited studies investigating the anticancer properties of these fragments. The fragment with improved anticancer activities is an attractive alternative to chemotherapeutic drugs—presenting severe side effects. Herein, we report the isolation and characterization of recombinant engineered-lactoferrin (rtHLF4), exhibiting up to 100-fold improved anticancer activity compared to the full-length lactoferrin (flHLF). Further, rtHLF4 exerts its anticancer effect in a shorter duration. Through transcriptomic analysis of various cancer biomarkers, rtHLF4 was found to upregulate various pro-apoptotic markers and downregulate signaling proteins involved in angiogenesis and metastasis. We further determined that rtHLF4 showed no hemolytic activity at high concentrations. We believe that this anticancer protein can be further developed as a cancer treatment.
Yu Pan, Niying Chua, Kaisheng Lim, and Chun Loong Ho, Engineering of Human Lactoferrin for Improved Anticancer Activity, ACS Pharmacology & Translational Science 20214 (5), 1476-1482, DOI: 10.1021/acsptsci.1c00134
Bovine Milk Lactoferrin Selectively Kills Highly Metastatic Prostate Cancer PC-3 and Osteosarcoma MG-63 Cells In Vitro (2018)
Prostate cancer and osteosarcoma are the second most common type of cancer affecting men and the fifth most common malignancy among adolescents, respectively. The use of non-toxic natural or natural-derived products has been one of the current strategies for cancer therapy, owing to the reduced risks of induced-chemoresistance development and the absence of secondary effects. In this perspective, lactoferrin (Lf), a natural protein derived from milk, emerges as a promising anticancer agent due to its well-recognized cytotoxicity and anti-metastatic activity. Here, we aimed to ascertain the potential activity of bovine Lf (bLf) against highly metastatic cancer cells. The bLf effect on prostate PC-3 and osteosarcoma MG-63 cell lines, both displaying plasmalemmal V-ATPase, was studied and compared with the breast cancer MDA-MB-231 and the non-tumorigenic BJ-5ta cell lines. Cell proliferation, cell death, intracellular pH, lysosomal acidification, and extracellular acidification rate were evaluated. Results show that bLf inhibits proliferation, induces apoptosis, intracellular acidification, and perturbs lysosomal acidification only in highly metastatic cancer cell lines. By contrast, BJ-5ta cells are insensitive to bLf. Overall, our results establish a common mechanism of action of bLf against highly metastatic cancer cells exhibiting plasmalemmal V-ATPase. This study opens promising perspectives for further research on the anticancer role of Lf, which ultimately will contribute to its safer and more rational application in the human therapy of these life-threatening cancers.
Guedes JP, Pereira CS, Rodrigues LR, Côrte-Real M. Bovine Milk Lactoferrin Selectively Kills Highly Metastatic Prostate Cancer PC-3 and Osteosarcoma MG-63 Cells In Vitro. Front Oncol. 2018 Jun 4;8:200. doi: 10.3389/fonc.2018.00200. PMID: 29915723; PMCID: PMC5994723.
Regression of prostate tumors after intravenous administration of lactoferrin-bearing polypropylenimine dendriplexes encoding TNF-α, TRAIL, and interleukin-12 (2017)
Abstract
The possibility of using gene therapy for the treatment of prostate cancer is limited by the lack of intravenously administered delivery systems able to safely and selectively deliver therapeutic genes to tumors. Given that lactoferrin (Lf) receptors are overexpressed on prostate cancer cells, we hypothesized that the conjugation of Lf to generation 3-diaminobutyric polypropylenimine dendrimer would improve its transfection and therapeutic efficacy in prostate cancer cells. In this study, we demonstrated that the intravenous administration of Lf-bearing DAB dendriplexes encoding TNFα resulted in the complete suppression of 70% of PC-3 and 50% of DU145 tumors over one month. Treatment with DAB-Lf dendriplex encoding TRAIL led to tumor suppression of 40% of PC-3 tumors and 20% of DU145 tumors. The treatment was well tolerated by the animals. Lf-bearing generation 3-polypropylenimine dendrimer is therefore a highly promising delivery system for non-viral gene therapy of prostate cancer.
Najla Altwaijry, Sukrut Somani, John A. Parkinson, Rothwelle J. Tate, Patricia Keating, Monika Warzecha, Graeme R. Mackenzie, Hing Y. Leung & Christine Dufès (2018) Regression of prostate tumors after intravenous administration of lactoferrin-bearing polypropylenimine dendriplexes encoding TNF-α, TRAIL, and interleukin-12, Drug Delivery, 25:1, 679-689, DOI: 10.1080/10717544.2018.1440666
Bovine Lactoferrin Induces Cell Death in Human Prostate Cancer Cells (2022)
Abstract
Bovine lactoferrin (bLf) is a multifunctional protein widely associated with anticancer activity. Prostate cancer is the second most frequent type of cancer worldwide. This study was aimed at evaluating the influence of bLf on cell viability, cell cycle progression, reactive oxygen species (ROS) production, and rate of apoptosis in the human prostate cancer cell line (DU-145). MTT assay and trypan blue exclusion were used to analyze cell viability. Morphological changes were analyzed through optical microscopy after 24 h and 48 h of bLf treatment. FITC-bLf internalization and cellular damage were observed within 24 h by confocal fluorescence microscopy. Cell cycle analyses were performed by flow cytometry and propidium iodide. For caspases 3/7 activation and reactive oxygen species production evaluation, cells were live-imaged using the high-throughput system Operetta. The cell viability assays demonstrated that bLf induces cell death and morphological changes after 24 h and 48 h of treatment compared to control on DU-145 cells. The bLf internalization was detected in DU-145 cells, G1-phase arrest of the cell cycle, caspase 3/7 activation, and increased oxidative stress on bLf-treated cells. Our data support that bLf has an important anticancer activity, thus offering new perspectives in preventing and treating prostate cancer.
Vanessa P. Rocha, Samir P. C. Campos, Caroline A. Barros, Pablo Trindade, Leticia R. Q. Souza, Triciana G. Silva, Etel R. P. Gimba, Anderson J. Teodoro, Rafael B. Gonçalves, "Bovine Lactoferrin Induces Cell Death in Human Prostate Cancer Cells", Oxidative Medicine and Cellular Longevity, vol. 2022, Article ID 2187696, 13 pages, 2022. https://doi.org/10.1155/2022/2187696
Lactoferrin may inhibit the development of cancer via its immunostimulatory and immunomodulatory activities (Review) (2022)
Abstract
Lactoferrin (Lf) is secreted by ectodermal tissue and has a structure similar to that of transferrin. Although Lf seems to be multifunctional, its main function is related to the natural defense system of mammals. The present review aims to highlight the major actions of Lf, including the regulation of cell growth, the inhibition of toxic compound formation, the removal of harmful free radicals and its important role in immune response regulation. Moreover, Lf has antibacterial, antiviral, antioxidant, anticancer and anti‑inflammatory activities. In addition, the use of Lf for functionalization of drug nanocarriers, with emphasis on tumor‑targeted drug delivery, is illustrated. Such effects serve as an important theoretical basis for its future development and application. In neurodegenerative diseases and the brains of elderly people, Lf expression is markedly upregulated. Lf may exert an anti‑inflammatory effect by inhibiting the formation of hydroxyl free radicals. Through its antioxidant properties, Lf can prevent DNA damage, thereby preventing tumor formation in the central nervous system. In addition, Lf specifically activates the p53 tumor suppressor gene.
Pan, S., Weng, H., Hu, G., Wang, S., Zhao, T., Yao, X., Liao, L., Zhu, X., Ge, Y."Lactoferrin may inhibit the development of cancer via its immunostimulatory and immunomodulatory activities (Review)". International Journal of Oncology 59.5 (2021): 85.
Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis (2020)
Abstract
Patients with acute kidney injury (AKI) who survive the acute stage are at notable risk for chronic kidney disease (CKD) progression. There is no single therapy that can effectively prevent the AKI to CKD transition. Autophagy is a cytoplasmic component degradation pathway and has complex functions in several diseases, such as renal fibrosis. Previous research has shown that lactoferrin has important functions in antioxidant defense and other defense systems, protecting kidneys against various injuries. The present study investigated the effect of lactoferrin in protecting against the AKI to CKD transition. We identified 62 consensus genes with two-fold changes in clinical kidney tissues from AKI and CKD patients. Among the 62 overlay genes, the mRNA levels of LTF were significantly upregulated in the kidney tissues of AKI and CKD patients. Lactoferrin induced autophagy via the activation of the AMPK and inhibition of Akt/mTOR pathway in human kidney proximal tubular cells. Lactoferrin suppressed oxidative stress-induced cell death and apoptosis by augmenting autophagy. Lactoferrin has an antifibrotic role in human kidney tubular cells. In a mouse model of folic acid-induced AKI to CKD transition, treatment with lactoferrin recovered renal function and further suppressed renal fibrosis through the inhibition of apoptosis and the induction of autophagy. These findings identify lactoferrin as a potential therapeutic target for the prevention of the AKI to CKD transition.
Hsu YH, Chiu IJ, Lin YF, Chen YJ, Lee YH, Chiu HW. Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis. Pharmaceutics. 2020 May 8;12(5):434. doi: 10.3390/pharmaceutics12050434. PMID: 32397266; PMCID: PMC7284869.
OTHER NAME(S): Apolactoferrin, Bovine Lactoferrin, Human Lactoferrin, Lactoferrina, Lactoferrine, Lactoferrine Bovine, Lactoferrine Humaine, Lactoferrine Humaine Recombinante, Lactoferrines, Lactoferrins, Recombinant Human Lactoferrin
Overview
Lactoferrin is a protein in human milk, animal milk, and other bodily fluids. Colostrum, the first milk made after a baby is born, is higher in lactoferrin.
Lactoferrin helps regulate how well iron is absorbed into the body from the intestine. It also seems to protect against infections from bacteria, viruses, and fungi. The lactoferrin in breastmilk is thought to help protect breast-fed infants against infections..
People commonly use lactoferrin for low iron levels during pregnancy and for preventing blood infections (sepsis) in premature infants. It is also used for diarrhea, common cold, and many other conditions, but there is no good scientific evidence to support these other uses.
Uses & Effectiveness
Possibly Effective for
Low iron levels during pregnancy. Taking lactoferrin as a source of iron during pregnancy is about as effective as taking iron supplements by mouth and might be as effective as an iron injection given by a healthcare provider.
Blood infection (sepsis). Giving lactoferrin by mouth to premature infants might help prevent serious blood infections.
Likely InEffective for
Death of an unborn or premature baby. Taking lactoferrin does not seem to prevent the death of premature infants.
A serious intestinal disease in premature infants (necrotizing enterocolitis or NEC). Taking lactoferrin does not seem to prevent premature infants from developing NEC.
There is interest in using lactoferrin for a number of other purposes, but there isn't enough reliable information to say whether it might be helpful.
Side Effects
When taken by mouth: Lactoferrin is commonly consumed in foods. Consuming higher amounts of lactoferrin from cow's milk is possibly safe for up to one year. Human lactoferrin that is made from specially processed rice appears to be safe for up to 14 days. Taking doses higher than 7.2 grams daily seems to increase the risk of side effects, such as skin rash, loss of appetite, constipation, diarrhea, and nausea.
When applied to the skin: There isn't enough reliable information to know if lactoferrin is safe. It might cause skin irritation.
When administered into the vagina: There isn't enough reliable information to know if lactoferrin is safe or what the side effects might be.
Special Precautions and Warnings
When taken by mouth: Lactoferrin is commonly consumed in foods. Consuming higher amounts of lactoferrin from cow's milk is possibly safe for up to one year. Human lactoferrin that is made from specially processed rice appears to be safe for up to 14 days. Taking doses higher than 7.2 grams daily seems to increase the risk of side effects, such as skin rash, loss of appetite, constipation, diarrhea, and nausea.
When applied to the skin: There isn't enough reliable information to know if lactoferrin is safe. It might cause skin irritation.
When administered into the vagina: There isn't enough reliable information to know if lactoferrin is safe or what the side effects might be. Pregnancy: Lactoferrin is commonly consumed in foods. Lactoferrin is possibly safe when taken by mouth in doses of 200 mg daily during pregnancy. There isn't enough reliable information to know if lactoferrin vaginal tablets are safe to use when pregnant. Stay on the safe side and avoid use.
Breast-feeding: Lactoferrin is commonly consumed in foods. But there isn't enough reliable information to know if taking larger amounts used as medicine by mouth or as vaginal tablets are safe when breast-feeding. Stay on the safe side and stick to food amounts.
Children: In infants and young children, lactoferrin is possibly safe when added to formula or other foods, such as yogurt. There isn't enough reliable information to know if lactoferrin is safe to use in children over 6 years old.
Interactions
We currently have no information for LACTOFERRIN Interactions.
Dosing
Lactoferrin has most often been used by adults in doses of 100-400 mg by mouth daily for up to 12 weeks. It's also used in topical and vaginal products. Speak with a healthcare provider to find out what type of product and dose might be best for a specific condition.
Repurposing of the antibiotic Doxycycline as an antiviral and anti-cancer therapeutic. A literature review (2022)
Abstract
This review begins with a brief introduction to doxycycline as a drug: its uses, side effects, history and chemical formula with the 5-ringed structure typical of the tetracyclines.
Doxycycline and its iron chelation mechanisms and effects are then described.
In 2021, Faure et al performed in vitro experiments to investigate the synergistic interactions between five tetracyclines and tobramycin with an iron chelator (CP762) against two reference strains and nine clinical isolates of Pseudomonas aeruginosa from cystic fibrosis patients.
They found that as it binds with high affinity to iron this inhibited its antibacterial effects by competing with the magnesium binding site on the bacterial ribosome. The addition of another iron chelator, CP762, synergistically restored the magnesium bridge binding.
In 1999 Alkawash et al appeared to find lactoferrin/doxycycline antibacterial synergy, and by a large margin of 32 to 64 fold against B. cepacia.
In 2015, Wu et al investigated its effects in vitro on the replication of vesicular stomatitis virus.
Of particular note here for also treating long covid/vaccine sequalae is that doxycycline acts as both an antiviral and an anticancer therapeutic agent by the induction of expression of the key tumor suppressor p53.
In 2020, Mosquera-Sulbaran and Hernández-Fonseca published a review on the use of tetracycline as an anti COVID-19 therapeutic.
2 clinical trials using doxycycline and ivermectin are then discussed. No results were available from the first of these, but from the second the only participants to die of COVID-19 were 3 from the placebo group of 200. Duration and severity of symptoms in the treatment group were also significantly reduced.
Three papers investigating iron chelation, inhibition of tumors and metastasis are reviewed. Buss et al (2003) recognized the potential of using iron chelation in cancer therapy and their possible synergistic effects.
From 2013, Richardson et al review how the iron chelator DFO can inhibit key signalling pathways which induce epithelial mesenchymal transition (EMT) in pancreatic cancer and other tumors. EMT is described.
A paper by Morales and Xue (2021) reviews the targeting of iron metabolism in cancer therapy.
Ubiquitination is described, as is evidence for the HIF inhibitory effects of both lactoferrin and ivermectin. This is important for working synergistically with doxycycline to help avoid resistance from cancer cells.
To complete this review, four papers discuss the anticancer properties of doxycycline.
In 1998, Fife et al found that, in vitro, doxycycline can significantly inhibit the growth of prostate and breast cancer tumors by the inhibition of matrix metalloproteinases (MMPs) and induction of apoptosis.
From 2016, Zhang et al conducted an in vitro study using human breast cancer cell lines.
A paper by Zhu et al (2017) conducted an in vitro investigation into how doxycycline synergizes with the chemotherapeutic drug doxorubicin to inhibit the proliferation of castration-resistant prostate cancer cells, a condition that was previously untreatable.
And from 2019, Markowska et al conducted a review into the repositioning of doxycycline, salinomycin, monensin and ivermectin as cancer drugs.
To conclude this Substack, dosing and contraindications for doxycycline monohydrate are considered.
Added 23rd December ‘23:
Caution is warranted as doxycycline is contraindicated for some patients, which is by no means unusual:
Doxycycline Promotes Carcinogenesis & Metastasis via Chronic Inflammatory Pathway: An In Vivo Approach (2016)
Artemisinin Blocks Prostate Cancer Growth and Cell Cycle Progression by Disrupting Sp1 Interactions with the Cyclin-dependent Kinase-4 (CDK4) Promoter and Inhibiting CDK4 Gene Expression (2009)
Abstract
Artemisinin, a naturally occurring component of Artemisia annua, or sweet wormwood, is a potent anti-malaria compound that has recently been shown to have anti-proliferative effects on a number of human cancer cell types, although little is know about the molecular mechanisms of this response. We have observed that artemisinin treatment triggers a stringent G1 cell cycle arrest of LNCaP (lymph node carcinoma of the prostate) human prostate cancer cells that is accompanied by a rapid down-regulation of CDK2 and CDK4 protein and transcript levels. Transient transfection with promoter-linked luciferase reporter plasmids revealed that artemisinin strongly inhibits CDK2 and CDK4 promoter activity. Deletion analysis of the CDK4 promoter revealed a 231-bp artemisinin-responsive region between -1737 and -1506. Site-specific mutations revealed that the Sp1 site at -1531 was necessary for artemisinin responsiveness in the context of the CDK4 promoter. DNA binding assays as well as chromatin immunoprecipitation assays demonstrated that this Sp1-binding site in the CDK4 promoter forms a specific artemisinin-responsive DNA-protein complex that contains the Sp1 transcription factor. Artemisinin reduced phosphorylation of Sp1, and when dephosphorylation of Sp1 was inhibited by treatment of cells with the phosphatase inhibitor okadaic acid, the ability of artemisinin to down-regulate Sp1 interactions with the CDK4 promoter was ablated, rendering the CDK4 promoter unresponsive to artemisinin. Finally, overexpression of Sp1 mostly reversed the artemisinin down-regulation of CDK4 promoter activity and partially reversed the cell cycle arrest. Taken together, our results demonstrate that a key event in the artemisinin anti-proliferative effects in prostate cancer cells is the transcriptional down-regulation of CDK4 expression by disruption of Sp1 interactions with the CDK4 promoter.
Prostate cancer is the most diagnosed cancer and the second leading cause of cancer death among men in the United States (1). One third of all cancer cases reported in men are prostate cancer, and one out of every six men will be diagnosed with prostate cancer at some point in their lifetimes (1). The primary treatment for patients diagnosed with prostate cancer is androgen ablation therapy, which consists of administering anti-androgens and chemical castration to decrease the levels of circulating androgens, such as testosterone, in the body (2). Given that prostate cancers initially develop as androgen-responsive, this ablation therapy is particularly effective early on in the course of treatment (3). However, androgen ablation treatment is associated with a 70–80% progression rate into androgen-independent prostate tumors within 1–3 years so despite the initial success of this therapy, in most cases, the cancer will relapse as an incurable hormone-refractory condition in which the overall survival is ∼15–16 months (4, 5). The lack of therapeutics that are highly effective against all types of prostate cancer is a critical problem in the field.
Naturally occurring plant compounds represent a largely untapped source of potential chemotherapeutic molecules to control different types of prostate cancers with very minimal side effects. One such promising compound is artemisinin, a sequiterpene lactone that was isolated from Artemisia annua (more commonly known as qinghaosu or sweet wormwood). Chinese medical practitioners have used artemisinin from plant extracts for over two thousand years to treat a variety of conditions such as fever and hemorrhoids. The compound was isolated from A. annua by Chinese chemists in the 1970s, and since then, artemisinin and a number of its derivatives have been used to effectively treat different forms of malaria (6). Recently, artemisinin and its derivatives have been shown to induce growth arrest and apoptosis (7–9), as well as inhibit angiogenesis by down-regulation of the vascular endothelial growth factor vascular epidermal growth factor and its cellular receptor KDR/flk-1 (10, 11). One study that analyzed 55 cell lines of the Developmental Therapeutics Program of NCI, National Institutes of Health, showed that artesunate, the semisynthetic derivative of artemisinin, has anti-cancer activities against leukemic, colon, melanoma, breast, ovarian, prostate, central nervous system, and renal cancer cell lines (12). Moreover, the highly stable artemisinin-derived trioxane dimmers was shown to inhibit the growth of and selectively kill several human cancer cell lines without inducing cytotoxic effects on normal neighboring cells (13). The molecular mechanism and gene expression changes that mediate the anti-proliferative activity of artemisinin are not well characterized.
Eukaryotic cell growth depends on the cooperative actions of a number of cellular proteins to form a series of regulated events that drive the cell cycle from one phase to the next. The cell cycle is composed of four phases: G1 phase, S phase, involving DNA synthesis, G2 phase, and mitosis, or M phase where the cell divides. Critical components of the cell cycle machinery are the cyclin-dependent kinases (CDKs),2 their activating binding partners called cyclins, and a variety of cyclin-dependent kinase inhibitors (CKIs). CDKs bind to specific cyclin subunits to achieve the kinase activity necessary for the phosphorylation of substrates needed for the progression of the cell cycle, such as retinoblastoma (Rb) protein (14). In the G1 phase of the cell cycle, unphosphorylated Rb binds to the E2F family of transcription factors preventing them from activating the genes necessary for progression through S phase (15). Early in the G1 phase, CDK4 and CDK6, interacting with D-type cyclins, phosphorylate the Rb protein in an “initiation” step. In mid to late G1, CDK2 can then hyperphosphorylate the Rb protein by interacting with E-type cyclins. The hyperphosphorylation of Rb causes the release the E2F transcription factor allowing the cell to enter S phase and begin DNA replication (15). The correct timing and regulation of the cell cycle is mediated through CDK activity by the control of cyclin stability, subcellular localization of the components, CDK phosphorylation events, and association of the CDKs with CKIs (16). In this study, we examine the affects of artemisinin on the LNCaP (lymph node carcinoma of the prostate) cell cycle and we have discovered that artemisinin regulates expression of key G1 acting CDKs through the selective control of Sp1 transcription factor-promoter interactions. The results implicate artemisinin as a potential chemotherapeutic compound for controlling the proliferation of human prostate carcinoma.
Willoughby JA Sr, Sundar SN, Cheung M, Tin AS, Modiano J, Firestone GL. Artemisinin blocks prostate cancer growth and cell cycle progression by disrupting Sp1 interactions with the cyclin-dependent kinase-4 (CDK4) promoter and inhibiting CDK4 gene expression. J Biol Chem. 2009 Jan 23;284(4):2203-13. doi: 10.1074/jbc.M804491200. Epub 2008 Nov 17. PMID: 19017637; PMCID: PMC2629082.
Glucosamine suppresses proliferation of human prostate carcinoma DU145 cells through inhibition of STAT3 signaling (2009)
Abstract
Background
Glucosamine is known as a toxic agent for several malignant cell lines and transplanted tumors with little toxicity to normal host tissues. However, the mechanisms underlying anticancer activity of glucosamine are poorly understood. To study the mechanisms, the human prostate cancer DU145 cells were used for the model.
Results
Glucosamine at concentration 2 mM suppressed proliferation and induced death of DU145 cells. Detailed analysis showed that glucosamine decreased DNA synthesis, arrested cell cycle at G1 phase and induced apoptosis. The effects of glucosamine were associated with up-regulation of p21waf1/cip, a CDK inhibitor. Our further studies identified glucosamine as an inhibitor of signal transducer and activator of transcription (STAT) 3 which is constitutively activated in many cancer cells including DU145 cells. Glucosamine inhibited phosphorylation of STAT3 at the Tyr705 residue and as a result, reduced STAT3 DNA binding and transcriptional activities. Indeed, the expression of apoptosis inhibitor survivin, which is well known target of STAT3, was suppressed. Contrary to DU145 cells, glucosamine did not affect proliferation of other human prostate cancer PC-3 and C4-2B cells, in which STAT 3 signal pathway is not constitutively active.
Conclusion
Our data identifies glucosamine as a suppressor of STAT3 signaling and suggests that anticancer activity of glucosamine may be attributed to the suppression of STAT3 activity. Potential application of glucosamine for the treatment of tumors with constitutively active STAT3 is proposed.
Chesnokov, V., Sun, C. & Itakura, K. Glucosamine suppresses proliferation of human prostate carcinoma DU145 cells through inhibition of STAT3 signaling. Cancer Cell Int9, 25 (2009). https://doi.org/10.1186/1475-2867-9-25
Subject to checks for intolerance or contraindications, I recommend taking berberine with milk thistle to increase bioavailability. Milk thistle itself also has a great anticancer profile.
Metabolic effect of berberine-silymarin association: A meta-analysis of randomized, double-blind, placebo-controlled clinical trials (2019)
Abstract
The aim of this study is to assess the impact of a combination of berberine and silymarin on serum lipids and fasting plasma glucose (FPG) through a systematic review of literature and meta-analysis of the available randomized, double-blind, placebo-controlled clinical trials (RCTs). A systematic literature search in SCOPUS, PubMed-Medline, ISI Web of Science, and Google Scholar databases was conducted up to October 2, 2018, in order to identify RCTs assessing changes in plasma concentrations of total cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and FPG during treatment with berberine and silymarin in combination. Two review authors independently extracted data on study characteristics, methods, and outcomes. Quantitative data synthesis was performed using a random-effects model. We identified five eligible RCTs, with 497 subjects overall included. Berberine and silymarin combination treatment exerted a positive effect on TC (mean difference [MD]: -25.3, 95% CI [-39.2, -11.4] mg/dl; p < 0.001), TG (MD: -28, 95% CI [-35.3, -20.6] mg/dl; p < 0.001), HDL-C [MD: 6, 95% CI [3.2, 8.8] mg/dl; p < 0.001), LDL-C (MD: -29.1, 95% CI [-39.7, -18.6] mg/dl; p < 0.001), and FPG (MD: -7.5, 95% CI [-13, -1.9] mg/dl; p = 0.008). The present findings suggest that the coadministration of berberine and silymarin is associated with an advantageous improvement in lipid and glucose profile, suggesting the possible use of this nutraceutical combination in order to promote the cardiometabolic health.
Silymarin (milk thistle). An anticancer therapeutic in its own right, amongst other benefits, I take it with many agents to improve bioavailability eg Q10, quercetin, resveratrol and especially berberine. It also helps counter multidrug resistance, helping restore the actions of other chemotherapies.
Anticancer Potential of Silymarin: From Bench to Bed Side (2006)
Abstract
Silymarin consists of a family of flavonoids (silybin, isosilybin, silychristin, silydianin and taxifoline) commonly found in the dried fruit of the milk thistle plant Silybum marianum. Although silymarin's role as an antioxidant and hepatoprotective agent is well known, its role as an anticancer agent has begun to emerge. Extensive research within the last decade has shown that silymarin can suppress the proliferation of a variety of tumor cells (e.g., prostate, breast, ovary, colon, lung, bladder); this is accomplished through cell cycle arrest at the G1/S-phase, induction of cyclin-dependent kinase inhibitors (such as p15, p21 and p27), down-regulation of anti-apoptotic gene products (e.g., Bcl-2 and Bcl-xL), inhibition of cell-survival kinases (AKT, PKC and MAPK) and inhibition of inflammatory transcription factors (e.g., NF-κB). Silymarin can also down-regulate gene products involved in the proliferation of tumor cells (cyclin D1, EGFR, COX-2, TGF-β, IGF-IR), invasion (MMP-9), angiogenesis (VEGF) and metastasis (adhesion molecules). The anti-inflammatory effects of silymarin are mediated through suppression of NF-κB-regulated gene products, including COX-2, LOX, inducible iNOS, TNF and IL-1. Numerous studies have indicated that silymarin is a chemopreventive agent in vivo against a variety of carcinogens/tumor promoters, including UV light, 7,12-dimethylbenz(a)anthracene (DMBA), phorbol 12-myristate 13-acetate (PMA) and others. Silymarin has also been shown to sensitize tumors to chemotherapeutic agents through down-regulation of the MDR protein and other mechanisms. It binds to both estrogen and androgen receptors, and down-regulates PSA. In addition to its chemopreventive effects, silymarin exhibits antitumor activity against human tumors (e.g., prostate and ovary) in rodents. Various clinical trials have indicated that silymarin is bioavailable and pharmacologically safe. Studies are now in progress to demonstrate the clinical efficacy of silymarin against various cancers.
RAJESH AGARWAL, CHARU AGARWAL, HARUYO ICHIKAWA, RANA P. SINGH, BHARAT B. AGGARWAL, Anticancer Potential of Silymarin: From Bench to Bed Side, Anticancer Research Nov 2006, 26 (6B) 4457-4498;
“Zinc has been found to inhibit prostate cancer cell line growth and invasion. In part, this may be through the inhibition of nuclear factor κB, an antiapoptotic protein. Tissue levels of zinc are consistently lower in prostate cancer specimens when compared with normal specimens.”
But do not exceed 80mg/day or it may become counterproductive. They don’t say, but this may be due to copper deficiency:
Does Zinc Supplementation Increase the Risk of Prostate Cancer? (2005)
In the United States, prostate cancer is the most commonly diagnosed cancer in men and the second leading cause of cancer deaths in this population. It is estimated that 220 000 cases of prostate cancer will be diagnosed this year, and this is expected to increase with the expanding geriatric population. The etiology of prostate cancer is multifactorial. Genetic factors are important and contribute to incidence rates that are higher in African Americans than any other racial group. It is also clear that diet plays an important role in modulating the cancer phenotype. There is expanding interest, both in the lay press and the scientific community, in the use of dietary supplements that minimize the initiation and progression of prostate cancer.
The concentration of zinc in the prostate is higher than that of any other soft tissue in the body. Zinc is a necessary component of numerous metalloproteins including those important for DNA synthesis, immune function, and antioxidant activity. It is estimated that 15% of the population uses zinc supplements to exceed the recommended dietary allowance of 11 mg/d for men.1 A large body of literature supports a protective role for zinc with regard to prostate cancer progression. Zinc has been found to inhibit prostate cancer cell line growth and invasion.2 In part, this may be through the inhibition of nuclear factor κB, an antiapoptotic protein.3 Tissue levels of zinc are consistently lower in prostate cancer specimens when compared with normal specimens.4,5 This may be due to the down-regulation or inactivation of zinc transporter proteins,6 a finding also hypothesized to contribute to increased disease in African Americans.7 Low serum levels of zinc are associated with an increased incidence of prostate cancer,8-10 although these findings have been contradicted.11 In recent case-control studies, a weak to moderate protective effect of higher zinc intake was found against prostate cancer.12,13 Objective data analyzing zinc levels in toenail clippings demonstrate no statistical difference between patients with prostate cancer and controls and a moderate protective effect for higher zinc concentrations.14 These data suggest that the risk of prostate cancer may be lower among men with a moderate to higher zinc intake.
However, a recent study involving 46 000 health professionals (the Health Professionals Follow-up Study)15 found that men who consumed more than 100 mg/d of supplemental zinc had a higher relative risk (2.9-fold) of advanced prostate cancer. This increase in risk was amplified with the long-term intake of zinc supplements for more than 10 years. Supplemental zinc provided 32% of the total zinc intake and was the major source. The reasons behind the increased risk with zinc intake found in this study are unclear. At extremely high levels, more than 150 mg/d, zinc may cause immune dysfunction.16 Zinc is also correlated with higher levels of circulating insulinlike growth factor I, which are related to prostate cancer development.17 The men in the Health Professionals Follow-up Study who consumed supplemental zinc also consumed increased levels of other supplements, notably calcium, and were less likely to have a history of prostate cancer screening, both potentially confounding factors. The correlation was found only with advanced cancers (ie, with local extension or metastases) and not with organ-confined cancers, which represent the vast majority of diagnosed cancers. This suggests that the effect, if any, may be in the promotion of aggressive cancers. Only 10 of 2127 patients diagnosed as having prostate cancer during the study had advanced cancer and a zinc intake of greater than 100 mg/d. In sum, the health professionals data suggest that high levels of supplemental zinc may contribute to prostate cancer promotion in a small minority of patients. Further validation of this effect is needed.
Because of the Health Professionals Follow-up Study,15 concern has been generated regarding the use of supplemental zinc in aging men, a group at high risk for prostate cancer development. The recent Age-Related Eye Disease Study (AREDS)18 found that 80 mg/d of zinc, either alone, as zinc oxide, or as antioxidants and zinc, significantly reduced the risk of progression of age-related macular degeneration (AMD). Age-related macular degeneration is the leading cause of legal blindness in individuals 65 years or older and affects 1.7% to 1.9% of the population. In the AREDS study, zinc appeared to have a synergistic effect in combination with antioxidants. Prior to this study, no proven treatment had prevented or slowed the development of AMD for the 640 000 or more individuals with signs of late AMD.
If zinc supplementation becomes more widely used, should we be concerned about the development or progression of prostate cancer in these individuals? The levels of zinc supplementation used in the AREDS study were lower (80 mg) than those identified in the health professionals study as being a risk factor for prostate cancer. In addition, no increase in prostate cancer was noted in the patients receiving zinc supplementation in the former study. Given the known benefit of zinc for AMD, it is reasonable to prescribe zinc supplementation to patients at risk for progression to AMD. However, several caveats should be noted pending further data. Because of the increased risk of prostate cancer identified in patients with a supplemental intake of more than 100 mg/d, patients using the AREDS protocol should be warned not to take additional zinc. The routine intake of foods containing high amounts of zinc, such as beef and breakfast cereals, should be monitored. In addition, the consumption of other supplements, especially those containing calcium, should be minimized or avoided. Given the uncertainty regarding the effect of zinc on cancer progression, it may also be reasonable to avoid zinc supplementation in men diagnosed as having prostate cancer or precancerous lesions of the prostate such as high-grade prostatic intraepithelial neoplasia. One interesting aspect of the AREDS study was the finding that in individuals receiving zinc supplementation, there was an increase in hospitalization for genitourinary symptoms (8.6% vs 4.4%; P<.001). The reasons for this difference are not clear and will require further study. At this point, lower urinary tract symptoms should not be considered a contraindication.
In conclusion, 80 mg/d of supplemental zinc for the prevention of AMD does not appear to significantly increase the risk of prostate cancer in most patients. A significant body of data suggests that zinc may play a role in inhibiting prostate cancer at the lower levels used in the AREDS study. Given the known morbidity of AMD and the beneficial impact of zinc on this disease, the use of zinc supplementation should be encouraged in elderly patients at risk for macular degeneration
Jarrard DF. Does Zinc Supplementation Increase the Risk of Prostate Cancer? Arch Ophthalmol. 2005;123(1):102–103. doi:10.1001/archopht.123.1.102
Induction of apoptosis in prostate cancer cell lines by a flavonoid, baicalin (2000):
Prostate cancer is one of the most common types of cancer in men. Usually prostate cancer grows slowly and is initially confined to the prostate gland, where it may not cause serious harm. However, while some types of prostate cancer grow slowly and may need minimal or even no treatment, other types are aggressive and can spread quickly. Prostate cancer that's detected early — when it's still confined to the prostate gland — has the best chance for successful treatment.
In 2000, Chan et al conducted an in vitro study into the effects of baicalin on several prostate cancer cell lines. They found that the responses to baicalin were different among different cell lines, and that 50% inhibition of DU145 cells (considered a standard prostate cancer cell line) occurred at concentrations of 150μmol or above. LNCaP cells were the most resistant (androgen-sensitive human prostate adenocarcinoma cells):
Abstract
The flavonoid baicalin (baicalein 7-D-beta-glucuronate), isolated from the dried root of Scutellaria baicalensis Georgi (Huang Qin), is widely used in the traditional Chinese herbal medicine for its anti-inflammatory, anti-pyretic and anti-hypersensitivity effects. In the present study, we investigated the in vitro effects of baicalin on the growth, viability, and induction of apoptosis in several human prostate cancer cell lines, including DU145, PC-3, LNCaP and CA-HPV-10. The cell viability after treating with baicalin for 2-4 days was quantified by a colorimetric 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-s ulfophenyl)- 2H-tetrazolium (MTS) assay. The results showed that baicalin could inhibit the proliferation of prostate cancer cells. The responses to baicalin were different among different cell lines, with DU145 cells being the most sensitive and LNCaP cells the most resistant. Baicalin caused a 50% inhibition of DU145 cells at concentrations of 150 microM or above. The inhibition of proliferation of prostate cancer cells after a short period of exposure to baicalin was associated with induction by apoptosis, as evidenced by the typical nuclear fragmentation using Hoechst 33258 staining, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) labeling, DNA fragmentation, activation of caspase-3 and cleavage of poly-ADP-ribose polymerase (PARP). The results indicate that baicalin has direct anti-tumor effects on human prostate cancer cells.
The absorbance of the baicalin-treated cells was expressed as a percentage of the control untreated cells in order to show the extent of inhibition on cell growth (Fig. 2).
Fig. 2. Effect of baicalin on the proliferation of several human prostatic cancer cell lines. The cells were treated with increasing concentrations of baicalin (25–800 μM) for 4 days. Viable cells were measured by an MTS assay and expressed as a percentage of the controls (n=4). Standard errors are shown as vertical bars. Asterisks indicate that the levels are significantly different from the untreated controls (*P<0.05).
The cytotoxicity of baicalin on different cell lines was also assessed by fluorescent labeling of viable and dead cells with the calcein AM and EthD-1 (Fig. 3).
Fig. 3. Simultaneous fluorescent labeling of viable and non-viable DU145 cells using calcein-AM and EthD-1. DU145 cells were: (a), untreated (control treatment); or (b), treated with 400 μM baicalin for 4 days. (a) The cytoplasms of the viable cells in the control treatment were stained with calcein-AM; whereas (b), the nuclei of the non-viable baicalin-treated cells (indicated by arrows) were stained with EthD-1. A few viable cells (indicated by arrowheads in (b)) were also stained with calcein-AM in the baicalin-treated cells. Magnification, ×400.
The characteristic fragmented condensed nuclei or apoptotic bodies were clearly shown in apoptotic cells after staining with Hoechst 33258 (Fig. 4):
Fig. 4. Fluorescent staining of nuclei in baicalin-treated DU145 and LNCaP cells by Hoechst 33258. Cells were treated with 400 μM baicalin for 3 days. Apoptotic cells with fragmented nuclei (arrows) are seen in the baicalin-treated cells: (b), DU145; and (c), LNCaP; but not in the control treatment: (a), DU145. Magnification, ×400.
Apoptotic DU145 and LNCaP cells were also demonstrated by fluorescent TUNEL labeling and the positive apoptotic cells clearly exhibited the fluorescent labeled fragmented nuclei (Fig. 5a,b):
Fig. 5. Fluorescent TUNEL labeling of baicalin-treated apoptotic prostatic cancer cells. (a) DU145; and (b), LNCaP cells were treated with 400 μM baicalin for 3 days before TUNEL labeling. Positive apoptotic cells exhibit the characteristic fragmented nuclei. More apoptotic cells appeared in the LNCaP cells than in the DU145 cells after 3 days of baicalin treatment. Magnification, ×400.
There have been several in vitro studies showing that purified baicalin, or an extract from a Japanese medicinal herbal preparation (Sho-saiko-to for treating various chronic liver diseases) which is rich in both baicalin and baicalein, has demonstrated some anti-proliferative or anti-tumor effects on several human hepatoma cell lines, a pancreatic cancer cell line and a cholangiocarcinoma cancer line, rat glioma cells, a transgenic mouse model of melanoma and a derived melanoma cell line from this transgene. It has also been shown that baicalin is more potent than baicalein (which does not carry a glucuronate group) on the human hepatoma cells, and it takes 20 μg/ml of baicalin to inhibit growth of PLC/PRF/5 hepatoma cells by 50%. On the other hand, the herbal medicine, Sho-saiko-to, was shown to have no suppressive effects on the normal human peripheral blood lymphocytes and normal rat hepatocytes. It was also demonstrated that baicalin can prevent the formation of aberrant crypt foci induced by azoxymethane in the rat colon by inhibiting the cyclooxygenase activity, suggesting that this drug could be used as a chemopreventive agent for colon cancer.Recently, a herbal preparation, named PC-SPES, has been sold in the US as a dietary supplement for prostate cancer patients. This preparation, which contains extract from the S. baicalensis Georgi, has been claimed to be cytotoxic against PC-3 and LNCaP cells by the induction of apoptosis. In animal studies, this herbal preparation can also inhibit the growth and metastasis of a Dunning rat prostate cancer cell line, MAT-LyLu, grown in Copenhagen rats. In a recent clinical study, PC-SPES was shown to decrease the serum levels of testosterone and prostate-specific antigen in patients with hormone-sensitive prostate cancer.
In summary, our present study has shown that baicalin is cytotoxic for several human prostatic cancer cell lines, particularly the androgen-independent DU145 cells, and its inhibitory effect on these cells is mediated through the induction of apoptosis. Although its exact mechanisms on the induction of apoptosis on these cancer cells are not fully understood, baicalin could be promising as a chemopreventive agent or an adjuvant to the conventional therapeutic modalities for prostate cancers.
Acknowledgements
This study was supported partially by RGC Earmarked Research Grants, Hong Kong. The authors gratefully acknowledge Mrs HJ Chan-Hou for the confocal microscopy.
Chan FL, Choi HL, Chen ZY, Chan PS, Huang Y. Induction of apoptosis in prostate cancer cell lines by a flavonoid, baicalin. Cancer Lett. 2000 Nov 28;160(2):219-28. doi: 10.1016/s0304-3835(00)00591-7. PMID: 11053652.
As this has been heavily targeted I will provide some background and a couple of studies & links to do your own research. I cannot comment further for the same reasons.
From WebMD:
Is GcMAF a Potential Cancer Treatment? (2022)
What Is GcMAF?
GcMAF is short for "Gc protein-derived macrophage-activating factor." It's a type of immunotherapy, a treatment that revs up the immune system -- your body's defense against germs -- to kill cancer.
Macrophages are white blood cells the immune system sends out to gobble up foreign cells like bacteria and cancer. Our bodies make the protein GcMAF to activate macrophages. But cancer cells are thought to release an enzyme called nagalase that blocks the making of GcMAF to protect themselves against attack.
GcMAF treatment aims to activate more macrophages so they can fight cancer.
GcMAF Research
A 1997 study tested GcMAF on mice with cancer. It found that GcMAF improved their survival from 16 days to 32 days.
A few years later, the researchers tested the treatment on people with breast, colorectal, and prostate cancers. They gave them shots of a tiny amount of GcMAF once a week. After a few months, all of the patients were cured, according to the studies. Four to 7 years later, their cancers hadn't come back.
These results sound impressive, but there were some big problems with the studies. For one thing, they were very small -- just eight to 16 people each. Everyone in the studies had already been on standard cancer treatments like surgery, chemotherapy, or radiation. So it was hard to tell whether these treatments, or GcMAF, caused the cancers to shrink.
Also, doctors usually use imaging and lab tests to stage cancers -- in other words, to see how big the cancer is and whether it has spread. The researchers didn't do this. Instead, they took blood tests to check nagalase levels, which isn't a proven way to check for cancer or to see if it has gotten smaller.
Finally, the researchers never tested whether GcMAF actually activated macrophages in the patients' blood. So they couldn't be sure that the treatment was working at all.
Three doctors from the Anticancer Fund, a nonprofit group that promotes cancer research, published a letter in 2014 that outlined many of the concerns with the studies. They found several mistakes in the studies' claims and said that its conclusions "make no sense."
Future of GcMAF
A few researchers are still investigating GcMAF as a possible cancer treatment. Some early studies suggest that it may be helpful for people with late-stage cancers.
It's hard to know whether GcMAF works. The studies that have been done so far looked at very small numbers of people. Some of them included only one person. Larger studies are needed to prove that this treatment works on cancer and that it's safe.
Macrophages may still hold promise. Researchers are trying to learn whether monoclonal antibodies or other drugs might help macrophages kill cancer cells.
Until we know more, doctors stick to other immunotherapies, like checkpoint inhibitors, that have more evidence behind them. If you have questions about GcMAF or any other cancer treatment you've read about online, your cancer doctor is the best person to answer them.
GcMAF and the Persecution of David Noakes, Lyn Thyer & Immuno Biotech (2021)
Recently business man David Noakes was released from prison having served six months following his conviction on four charges relating to the manufacture, sale and supply of an unlicensed medicine.
(This article originally published on in-this-together.com. Republished in full with permission.)
Noakes pled guilty to all charges, including one of money laundering. This is something the MHRA and the mainstream media (MSM) have been very keen to highlight because it casts Noakes as a ‘real criminal.’
Money laundering is an automatically levied charge if anyone ever sells an unlicensed ‘medication.’ Pleading guilty to selling an unlicensed medication automatically makes you guilty of so called ‘money laundering.’ David Noakes is no BCCI executive.
Over 6 years Immuno Biotech made £7.6 million selling GcMAF. Out of that they paid a staff team of 27 including 4 research scientists, 7 doctors, 2 ultrasound staff, 4 nurses and admin staff for 6 years. They paid for the laboratories, staff travel (a significant expense) and accommodation. Any additional revenue they pumped back into GcMAF research and development. The CEO of GlaxoSmithKline earns approximately £6 million every single year.
The alleged medicine is not a synthetic manufactured pharmaceutical. It is actually derived from naturally occurring human protein. It is called ‘Gc Protein-derived Macrophage Activating Factor,’ or GcMAf for short. How and why GcMAF is being withheld from the public, despite an abundance of supporting scientific evidence, reveals a system of corrupt corporate control designed to profit from our sickness and death.
The scientific evidence clearly shows that GcMAF is potentially the most effective cancer treatment ever discovered. At David Noakes trial Judge Nicholas Lorraine-Smith made it clear that GcMAF was not on trial. He accepted that Noakes had acted out of a genuine desire to treat people; he noted that GcMAF had been instrumental in successfully treating people who had been written off by the medical profession and added that he was looking forward to GcMAF being made available to the public. He then sentenced David Noakes to prison.
Judge Nicholas Loraine Smith had little choice, and was compelled to make the required legal decision. He clearly felt uncomfortable and gave David Noakes just 15 months instead of the fourteen year sentence the Medicines & Healthcare Products Regulatory Agency (MHRA) were seeking. The difficulty he faced was highlighted when he stated, during the trial, that the court was not a court of morality but rather a court of law.
Clearly, in this case, the law is an ass. If the UK state recognised the codified British constitution then a jury could have annulled this statutory lunacy. However, through 800 years of lies and deception, the UK Parliament has unconstitutionally seized illegitimate sovereignty for itself and its statute laws. It was under this corrupt system the MHRA brought the case against David Noakes. When asked at the trial if he would do the same again David Noakes looked the Judge in the eye and said he would. He is a man who commands considerable respect in my opinion.
Under the 1939 Cancer Act it is illegal in the UK to advertise any cancer treatment which is not approved by the state. It is also illegal to offer any claimed cancer treatment, prescribe any claimed cancer treatment or offer any non-state sanctioned cancer treatment advice. I am not medically qualified, am not offering any medical advice and am not promoting GcMAF. I recommend you always seek qualified medical advice if you are ill. Rather, I am exposing what seems to be a rank injustice and questioning the system of cancer treatment approval and regulation in the UK.
Promising role for Gc-MAF in cancer immunotherapy: from bench to bedside (2017)
Abstract
Immunotherapy has been used for years in many types of cancer therapy. Recently, cancer immunotherapy has focused on mechanisms which can enhance the development of cell-mediated immunity. Anticancer medications are administered to inhibit immunosuppressive factors such as nagalase enzyme, which is produced by neoplastic cells and destroys macrophage activating factor (Gc-MAF). Anti-neoplastics medications can also enhance immune-cell activity against tumors. Such medications show great potential in cancer immunotherapy using natural human mechanisms against neoplasms.
Key Words: Cancer, Immunotherapy, Macrophage activating factor, Gc-MAF, Vitamin D.
Function, biological and pharmacological properties of Gc-globulin and Gc-MAF: The vitamin D-binding protein (DBP) Gc-globulin (human group-specific component), in addition to the storage and transport of vitamin D, has an important physiological function as a scavenger of extracellular G-actin to increase neutrophil chemotaxis and macrophage activation (11). Studies have shown that Gc- globulin is a protein with a structure similar to albumin that is a receptor of active vitamin D3 (12). Gc- globulin plays a role in immune system regulation, osteoclastic activity and as a primary defense against infectious factors such as immunodeficiency virus and sepsis. Gc-globulin when modified is capable of affecting the activation and fortification of immune cells exhibiting anticancer activity. These molecules activate macrophages after deglycosylation through β-galactosidase and sialidase of the B and T lymphocytes, respectively. This product supports phagocyte, superoxidase and immunopotentiatory activity of Gc-MAF (group specific component-macrophage activating factor; fig 1) (11-13).
Macrophages activated by Gc-MAF offer different properties that are effective against a variety of cancers in human and animal models (14, 15). Moreover, phenotypes of Gc-globulin influence the MAF levels in serum (16) and activate mouse peritoneal macrophages (17). Previous studies have confirmed that Gc as a precursor to Gc-MAF is considerably non-specific or even completely deglycosylated in cancer patients (fig 2) (18-20).
Adjunct medications: In some studies, adjunct drugs (INF-α and anti-thrombin-3) or supplemental immunotherapy (NK/T cell therapy) has been used to increase the efficiency of Gc-MAF (36, 58). Gc-MAF can be effective in the prevention of lesion progression through accompanying factors like VEGF-A, FGF-2 and CD36 receptor-mediated angiopoietin (27, 28). The role of chemotherapy medications in conjunction with Gc-MAF therapy must be determined. Administration of Gc-MAF for cancer patients exclusively activates macrophages as an important cell in adaptive immunity. It has been demonstrated that Gc-MAF does not directly activate other immune cells, such as dendritic cells (35), but that contributing macrophages to processed antigens via MHC-II antigen complex-mediation to T-cells appear to predominantly engaged B cells (21). Gc-MAF supports humoral immunity by producing, developing and releasing large quantities of antibodies against cancer. Clinical evidence from a human model of breast cancer patients supports this hypothesis (36, 37). There is also evidence that confirms the tumoricidal role of Gc-MAF via Fc-receptor mediation (59).
When exploring the role of other types of chemotherapy in conjunction with Gc-MAF, the role of nutrition cannot be overlooked. A favorable PINI score is associated with prolonged survival of advanced cancer patients and it is logical to assume that cancer patients who can prevent the onset or presence of cachexia will have a much stronger response to immunotherapy with Gc-MAF (60). The complexity of changes to the immune system in response to chronic inflammation associated with cachexia is far greater than previously envisaged. It is likely that the best therapeutic responses will be observed when the nutritional and inflammatory aspects are taken together with stimulation of the immune system (61).
Previous clinical investigations have confirmed the efficacy of Gc-MAF. In addition to activating existing macrophages, Gc-MAF is a potent mitogenic factor that can stimulate the myeloid progenitor cells to increase systemic macrophage cell counts by 40-fold in four days (48). The recent availability of food-based Gc-MAF, which is Gc-MAF produced during the fermentation of milk products, can provide further fields of research and application of this promising approach (73).
Although Gc-MAF was successfully used for immunotherapy of cancer patients, their aspect should be considered for future research.
In conclusion activation and contribution of cells (NK and T helper lymphocytes) and factors relating to immunotherapy are more complicated and costly than using Gc-MAF therapy. Moreover, these cells and factors have shown about 10-fold lower potent activity than the naturally activated (inflammation-primed) macrophages. There is a need to design further studies to directly compare the efficacy of routine cancer immunotherapy using activating NK versus Gc-MAF therapy.
The question must also be posed as to why this medication has not yet been approved by the FDA. Despite the doubts raised as results of some clinical studies, the efficacy of this drug has been endorsed in several studies. It appears that there are non-scientific reasons that prevent FDA approval.
Acknowledgments
We would like to thank Dr. Ashraf Karbasi and Dr. Marco Ruggiero for their valuable comments.
Immunotherapy with GcMAF revisited - A critical overview of the research of Nobuto Yamamoto (2022)
Abstract
This overview describes the research of Nobutu Yamamoto (Philadelphia) concerning immunotherapy with GcMAF for patients with cancer and for patients infected with pathogenic envelope viruses. GcMAF (Group-specific component Macrophage-Activating Factor) is a mammalian protein with an incredible potency to directly activate macrophages. Since the late 1980s Yamamoto's investigations were published in numerous journals but in order to understand the details of his research, a minute survey of many of his patents was required. But even then, regrettably, a precise description of his experiments was sometimes lacking. This overview tries to summarize all of Yamamoto's research on GcMAF, as well as some selected more recent papers from other investigators, who tried to verify and/or reproduce Yamamoto's reports. In my opinion the most important result of the GcMAF research deserves widespread renewed attention: human GcMAF injections (100 ng per week, intramuscular or intravenous) can help to cure patients with a great variety of cancers as well as patients infected with pathogenic envelope viruses like the human immunodeficiency virus 1 (HIV-1), influenza, measles and rubella (and maybe also SARS-CoV-2). From Yamamoto's data it can be calculated that GcMAF is a near-stoichiometric activator of macrophages. Yamamoto monitored the progress of his immunotherapy via the serum level of an enzyme called nagalase (α-N-acetylgalactosaminidase activity at pH 6). I have extensively discussed the properties and potential catalytic site of this enzyme activity in an Appendix entitled: "Search for the potential active site of the latent α-N-acetylgalactosaminidase activity in the glycoproteins of some envelope viruses".
Immunotherapy for Prostate Cancer with Gc Protein-Derived Macrophage-Activating Factor, GcMAF (2008)
Abstract
Serum Gc protein (known as vitamin D(3)-binding protein) is the precursor for the principal macrophage-activating factor (MAF). The MAF precursor activity of serum Gc protein of prostate cancer patients was lost or reduced because Gc protein was deglycosylated by serum alpha-N-acetylgalactosaminidase (Nagalase) secreted from cancerous cells. Therefore, macrophages of prostate cancer patients having deglycosylated Gc protein cannot be activated, leading to immunosuppression. Stepwise treatment of purified Gc protein with immobilized beta-galactosidase and sialidase generated the most potent MAF (termed GcMAF) ever discovered, which produces no adverse effect in humans. Macrophages activated by GcMAF develop a considerable variation of receptors that recognize the abnormality in malignant cell surface and are highly tumoricidal. Sixteen nonanemic prostate cancer patients received weekly administration of 100 ng of GcMAF. As the MAF precursor activity increased, their serum Nagalase activity decreased. Because serum Nagalase activity is proportional to tumor burden, the entire time course analysis for GcMAF therapy was monitored by measuring the serum Nagalase activity. After 14 to 25 weekly administrations of GcMAF (100 ng/week), all 16 patients had very low serum Nagalase levels equivalent to those of healthy control values, indicating that these patients are tumor-free. No recurrence occurred for 7 years.
Figure 3. Time course study of GcMAF therapy of 16 prostate cancer patients with serum α-N-acetylgalactosaminidase (Nagalase) as a prognostic index.
Case report: A breast cancer patient treated with GcMAF, sonodynamic therapy and hormone therapy (2014)
Abstract
Gc protein-derived macrophage-activating factor (GcMAF) occurs naturally in the human body. It has various functions, such as macrophage activation and antitumor activities. Recently, immunotherapy has become an attractive new strategy in the treatment of cancer. GcMAF-based immunotherapy can be combined with many other therapies. Sonodynamic therapy (SDT) using low-intensity ultrasound is a novel therapeutic modality. Ultrasound has been demonstrated to activate a number of sonosensitive agents allowing for the possibility of non-invasive targeted treatment for both superficial and deep-seated tumors. The current case study demonstrates that GcMAF and SDT can be used in combination with conventional therapies in patients with metastatic cancer, especially where treatment options are limited due to factors such as toxicity. This case study also suggests a new concept of cancer treatment using local destruction of cancer tissue, in this case conducted with SDT, to be used in combination with GcMAF immunotherapy as a systemic treatment.
Keywords: GeMAF; Immunotherapy; SDT; breast cancer; case report; macrophage; sonodynamic therapy.
By the beginning of October 2013 the patient showed dramatic improvement of symptoms such as cough, back pain and edema of the right hand from the combination therapy with SDT, GcMAF and hormone therapy. Her axillary tumor (Figure 1A) decreased in size and disappeared completely.
Figure 1. Chest tomography of a 55-year-old female patient on 6th June 2013 showing a right axillary tumor (A), spinal metastases (B), intrapleural nodular tumor and right pleural effusion (C).
We highlight this case of a patient with terminal breast cancer having had good effects from SDT, GcMAF and hormonal therapy. It suggests that SDT and GcMAF can be used in combination with standard treatments, in particular targeted-therapies, with minimal toxicity and without negative effects on the immune system, to achieve better outcomes for patients with cancer. SDT, GcMAF and hormonal therapies are non-invasive, well-tolerated treatments that may be capable of controlling tumor progression by working synergistically. Furthermore, SDT and GcMAF may be capable of controlling tumor progression by inducing direct inflammatory necrosis inside tumors, producing antitumor immunity via antigen-presenting cells to prevent immune escape in a variety of deep and superficial tumors.
Utilizing these new approaches gives those of us who have been treating cancer good weapons that kill cancer cells selectively, efficiently, and by non-toxic and painless means. We are planning to further refine and improve our protocols with SDT and GcMAF.
Case Report: A Non-small Cell Lung Cancer Patient Treated with GcMAF, Sonodynamic Therapy and Tumor Treating Fields (2016)
Abstract
Background/aim: Macrophage activating factor (MAF)-based immunotherapy has a wide application for use in treating many diseases via macrophage activation. Sonodynamic therapy (SDT) using low-intensity ultrasound and tumor treating field (TTF) therapy are novel therapeutic modalities. SDT is usually combined with ozone therapy to improve local hypoxia within the tumor environment.
Case report: We treated a 77-year-old male diagnosed with non-small cell lung cancer ((NSCLC) stage 3B) using second-generation serum GcMAF and oral colostrum MAF-based immunotherapy combined with SDT, TTF and ozone therapies.
Results: This case report demonstrates that GcMAF, oral colostrum MAF, SDT, TTF and ozone therapy can be used for NSCLC without adverse effects.
Conclusion: This case report suggests a new concept of cancer treatment using local destruction of cancer tissue, in this case conducted with SDT and TTF therapy, to be used in combination with serum GcMAF and colostrum MAF immunotherapy as a systemic treatment.
In patients with advanced-stage NSCLC, the prognosis is poor with a median survival of eight months when treated with platinum-based chemotherapy (24). In this case, using a combination of GcMAF immunotherapy, SDT, TTF and ozone therapies, the tumor had not enlarged for 15 months and several of the patient's symptoms improved. We highlight this case of a patient with terminal NSCLC having had good effects by using serum GcMAF and oral colostrum MAF-based immunotherapy combined with SDT, TTF and ozone therapies. It suggests that this combined therapy can be used together with standard treatments, in particular targeted-therapies, with minimal toxicity and without negative effects on the immune system, to achieve better outcomes for patients with cancer. Furthermore, the combined therapy may be capable of controlling tumor progression by inducing direct inflammatory necrosis inside tumors, producing antitumor immunity via antigen-presenting cells to prevent immune escape in a variety of deep and superficial tumors. We are planning to further refine and improve our protocols with this combined therapy.
Figure 2. Chest contrast-enhanced computed tomography (CT) image data before and after treatment. A: Chest contrast-enhanced computed tomography (CT) horizontal plane of a 77-year-old male patient on 13th January 2014 showing a right intrapleural nodular tumor before starting treatment with GcMAF, STD/PDT, ozone therapy and TTF. B: Chest contrast-enhanced CT horizontal plane on 20th April 2015 showing a low-density area inside the tumor.
Conclusion
The combination of MAF-based immunotherapy and local cancer destruction therapy can play a central role in future treatments against certain human cancers.
The amazing power of colostrum and lactoferrin (2018)
Colostrum
Colostrum is the first type of milk produced by mammals. It is produced by the mammary glands just prior to birth and its role is the feed the new born (it tends to be substantially higher in fat and protein than normal milk) and to protect the new born (it contains antibodies). Colostrum is thus crucial to the baby and a lack of it can cause stunted growth and even life-long immune problems. This is why breast feeding mother’s milk is so crucial to child and even adult health.
Colostrum is now known to contain important nutrients.
It contains exactly the right prebiotic food, inulin in particular, to feed the bacteria already received by the infant when it passed through mother’s birth canal. These bacteria have a bias towards strains of the species Bifodobacterium, with some strains of Lactobacillus.
It contains further bacteria to stimulate the infant’s immune response.
The combination sets up a highly acid gut in the infant – a pH of 5.5 – designed to keep pathogens in check.
It contains immune cells such as lymphocytes, antibodies including immunoglobulins which strengthen the inner gut lining and kick start the immune system, and cytokines including interleukins, chemokines and tumour necrosis factor.
It contains components of the innate immune system, for example lactoferrin, lactoperoxidase and lysozyme. These provide the components of the immune system which are stimulated when the host is attacked.
Finally, it contains important growth factors.
Bovine colostrum , for example, has been shown to boost natural killer cells and immune function (1). It has even been shown to combat Clostridium difficile (2).
Lactoferrin
Lactoferrin is a multi-functional iron-binding glycoprotein found at between 7-10 times the level in colostrum, as milk produced at a later stage of baby’s development. It seems to help generate and regulate the bone marrow in the infant and boost the whole innate immune defence. For example, when macrophages were cultured with lactoferrin, they produced high levels of tumour necrosis factor (3).
How does this occur? Lactoferrin is a Macrophage Activating Factor (MAF), which are lymphokines controlling antigens on the surface of macrophages, resulting in cells that can attack and kill cancer tumours. (You may have heard of GcMAF – the Gc refers to the stimulation by a vitamin D binding protein).
Lactoferrin is used as ‘medicine’ – it is used for treating stomach and intestinal ulcers. It is used as an antioxidant, as an immune stimulant and an anti-ageing supplement.
It is known to stimulate levels of good bacteria in the gut, reduce levels of the bad, and prevents constipation and diarrhea. It is an important first line defence against pathogens, yeasts and even viruses (4).
It also regulates the way the body processes iron.
Lactoferrin has been shown to help in a number of ‘auto-immune’ diseases including colitis, Crohn’s, Rheumatoid Arthritis and lupus because it stimulates certain gut bacteria.
However, that well-known UK Health Journal, the Daily Mail, has covered several stories since 2009 of breast-feeding women giving some of their breast milk to fathers who had prostate cancer and it extending their lives quite significantly.
Fenbendazole is an imidazole derivative and an anthelmintic (anti-parasitic) agent, just as ivermectin is. It is marketed as a veterinary drug, so direct drug interactions checks with human medications are not possible on the usual websites.
From Plumb’s Veterinary Drug Handbook (2011)
Pharmacology/Actions
Fenbendazole is a methylcarbamate benzimidazole antiparasitic agent and has a broad spectrum of activity against a variety of pathogenic internal parasites. In susceptible parasites, benzimidazole mechanism of action is believed due to disrupting intracellular microtubular transport systems by binding selectively and damaging tubulin, preventing tubulin polymerization, and inhibiting microtubule formation. Benzimidazoles also act at higher concentrations to disrupt metabolic pathways within the helminth, and inhibit metabolic enzymes, including malate dehydrogenase and fumarate reductase. Benzimidazoles may be considered time-dependent antiparasitic agents.
Pharmacokinetics
Fenbendazole is only marginally absorbed after oral administration. The amount absorbed from the gut is apparently more associated with the solubility of the drug and not the dose given. In dogs, when doses ranging from 25–100 mg/kg were administered, the area-under-the-curves were similar. Bioavailability is increased in dogs when fenbendazole is administered with food. Fat content of food does not significantly alter bioavailability (McKellar et al. 1993).
After oral dosing in calves and horses, peak blood levels of 0.11 micrograms/mL and 0.07 micrograms/mL, respectively, were measured. Absorbed fenbendazole is metabolized (and vice-versa) to the active compound, oxfendazole (sulfoxide) and the sulfone. In sheep, cattle, and pigs, 44–50% of a dose of fenbendazole is excreted unchanged in the feces, and <1% in the urine.
Contraindications/Precautions/Warnings
Fenbendazole is not FDA-approved for use in horses intended for food purposes.
Adverse Effects
At usual doses, fenbendazole generally does not cause any adverse effects. Hypersensitivity reactions secondary to antigen release by dying parasites may occur, particularly at high dosages. Salivation, vomiting, and diarrhea may infrequently occur in dogs or cats receiving fenbendazole. Pancytopenia has been reported in one dog (Gary et al. 2004).
Single doses (even at exaggerated doses) are not effective in dogs and cats; must treat for at least 3 days.
Reproductive/Nursing Safety
Fenbendazole is considered safe to use in pregnant bitches and is generally considered safe to use in pregnancy for all species. It is the drug of choice for treating giardia in pregnant animals (Tams 2007). In a system evaluating the safety of drugs in canine and feline pregnancy (Papich 1989), this drug is categorized as in class: A (Probably safe. Although specific studies may not have proved the safety of all drugs in dogs and cats, there are no reports of adverse effects in laboratory animals or women.)
Overdosage/Toxicity
Fenbendazole is apparently well tolerated at doses up to 100X recommended. The LD50 in laboratory animals exceeds 10 grams/kg when administered PO. It is unlikely an acute overdosage would lead to clinical signs.
Drug Interactions
BROMSALAN FLUKICIDES (dibromsalan, tribromsalan; not available in the USA): Oxfendazole or fenbendazole should not be given concurrently with the bromsalan flukicides; abortions in cattle and death in sheep have been reported after using these compounds together
Chemistry/Synonyms
A benzimidazole anthelmintic, fenbendazole occurs as a white, crystalline powder. It is only slightly soluble in water.
Fenbendazole may also be known as: Hoe-881V, Panacur®, and Safe-Guard®.
Storage/Stability
Fenbendazole products should be stored at room temperature.
Dosage Forms/Regulatory Status
VETERINARY-LABELED PRODUCTS:
Fenbendazole Granules: 222 mg/gram (22.2%) in 0.18 oz and 1 g, 2 g, 4 g packets and 1 lb jars; Panacur® Granules 22.2% (Intervet); (Rx); Safeguard® Canine Dewormer (Intervet), (OTC). FDA-approved for use in dogs, large exotic cats (lions, etc.), and bears (black bears, polar bears, etc.)
Fenbendazole Granules: 222 mg/gram (22.2%); Panacur® Granules 22.2% (Intervet). (OTC). FDA-approved for use in horses not intended for food.
Fenbendazole Suspension: 100 mg/mL (10%); available in both equine and bovine labeled products; Panacur® Suspension (Intervet); (Rx). FDA-approved for use in horses (not intended for food) and cattle. Slaughter withdrawal = 8 days (cattle). Safe-Guard® Suspension (Intervet); (OTC). FDA-approved for use in beef and dairy cattle. Slaughter withdrawal at labeled doses = 8 days
Fenbendazole Paste: 100 mg/gram (10%); available in both equine and bovine labeled products and sizes. Panacur® Paste (Intervet); (OTC). FDA-approved for use in horses (not intended for food) and cattle. Slaughter withdrawal at labeled doses = 8 days (cattle). Safe Guard® Paste (Intervet); (OTC). FDA-approved for use in horses not intended for food and cattle. Slaughter withdrawal at labeled doses = 8 days; no milk withdrawal time at labeled doses.
Combination Products:
Fenbendazole 454 mg, Ivermectin 27 mcg, & Praziquantel 23 mg (2.16 g small chews) Chewable Tablets; Panacur Plus® Soft Chews (Intervet); (Rx). FDA-approved for use in adult dogs.
Fenbendazole 1.134 g, Ivermectin 68 mcg, & Praziquantel 57 mg (5.4 g large chews) Chewable Tablet; Panacur Plus® Soft Chews (Intervet); (Rx). FDA-approved for use in adult dogs.
HUMAN-LABELED PRODUCTS: None
Exceptional Repositioning of Dog Dewormer: Fenbendazole Fever (2022)
Abstract
Fenbendazole (FZ) is a benzimidazole carbamate drug with broad-spectrum antiparasitic activity in humans and animals. The mechanism of action of FZ is associated with microtubular polymerization inhibition and glucose uptake blockade resulting in reduced glycogen stores and decreased ATP formation in the adult stages of susceptible parasites. A completely cured case of lung cancer became known globally and greatly influenced the cancer community in South Korea. Desperate Korean patients with cancer began self-administering FZ without their physician's knowledge, which interfered with the outcome of the cancer treatment planned by their oncologists. On the basis of presented evidence, this review provides valuable information from PubMed, Naver, Google Scholar, and Social Network Services (SNS) on the effects of FZ in a broad range of preclinical studies on cancer. In addition, we suggest investigating the self-administration of products, including supplements, herbs, or bioactive compounds, by patients to circumvent waiting for long and costly FZ clinical trials.
Figure 1. Mechanism of action of fenbendazole (FZ) targeting tubulin. Tubulin is the leading molecular target of FZ, which selectively binds to the β-tubulin of microtubules to disrupt microtubular polymerization, promoting immobilization and the death of parasites. The figure was created using Biorender (https://biorender.com/) (accessed on 15 May 2022). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9600184/
6. Complete Cure for Cancer by Self-Administering FZ with Supplements
In August 2016, a businessman from Oklahoma, Joe Tippens, was diagnosed with small-cell lung cancer and underwent a clinical trial under the supervision of his oncologist. He was informed of a short life expectancy from 3 months to 1 year. However, a veterinarian recommended to try FZ with vitamin E supplements, cannabidiol (CBD) oil, and bioavailable curcumin while going through the clinical trial. A positron emission tomography (PET) scan after 3 months did not detect any cancer cells anywhere in his body. [38]. Tippens was the only patient cured of cancer among the 1100 patients included in that clinical trial. Tippens shared his success story through Social Network Services (SNS) via a closed group, ‘my cancer story rocks’ (33,900 members) [39], and also in his blog, Get Busy Living (Figure 2), and mentioned at least 60 known FZ success stories [40]. The blog has been read by thousands from 60 different countries, as mentioned in his blog (Figure 3). A protocol is also available on the website recommending FZ 222 mg (1 gm of Panacur™ or Safeguard™) daily for different types of cancer such as colorectal, colon, lung, pancreatic, and prostate cancers, melanoma, lymphoma, and glioblastoma.
9. Discussion
To accelerate anticancer drug development, agents in clinical use for different indications are screened for repurposing. The approval of such repurposed drugs may be expedited owing to the availability of preclinical and clinical data on pharmacokinetics, toxicities, and regimens [53]. The anticancer activities of several anthelmintics, owing to their microtubule disruption ability, have generated considerable interest. The synergism of BZs, particularly FZ, with many clinically approved anticancer drugs is advantageous in repurposing these drugs. In the veterinary field, pets are often prescribed human medicines, since those available for companion animals are insufficient to cover all animal diseases. Likewise, commercially available animal drugs might be utilized in human medicine as long as human clinical trials have been conducted. However, globally, patients with cancer use SNS to repurpose veterinary medicines as anticancer drugs, following dosage regimens provided by self-cured patients. Sources of medical information on SNS are often unproven, and it is challenging for nonmedical professionals to precisely select and filter complex medical information. Considerable effort is required to set the effective dosages of FZ for humans. Moreover, if a patient self-administers a veterinary dewormer with an established anticancer drug while participating in a clinical trial, the outcome of the clinical trial could be altered completely, resulting in huge economic and time losses. Therefore, the reason for the therapeutic or adverse effects of the trialed drug in the clinical trial remains equivocal, and oncologists are confounded by their lack of knowledge regarding the self-administration of the dewormer (FZ) by patients.
FZ began to gain popularity as a human anticancer drug in South Korea in September 2019. The serious consequences of such events will likely emerge soon in South Korea. As the number of success stories being published online increases, the uninformed self-administration of FZ will also continue to increase in South Korea. Any prohibition by the government or medical doctors will likely not be followed by desperate patients with cancer. The resulting situation would lead to possible global FZ self-administration by patients with cancer. Therefore, an alternative must be offered to relieve patients from the long and expensive wait for FZ clinical trials in humans.
Indeed, patients with cancer and their families desperately seek remedies to treat the disease, but administering drugs with limited or no human safety profiles is a concern for clinicians. However, there are many exceptions for investigational drugs in clinical trials. There is a plethora of evidence that several BZ carbamates, particularly FZ, show anticancer potential in vivo, in vitro, and in silico. The role of MT is well-characterized, and the mechanism of action of MT disruptors such as FZ is similar to that of major chemotherapeutic agents such as vinblastine and vincristine.Nevertheless, nonmedical professionals cannot make appropriate medical judgments regarding the best usage of FZ. Therefore, we argue in favor of FZ owing to its well-established pharmacokinetics, excellent toxicity profile, and low cost, and suggest that an initial assessment of patients by interdisciplinary researchers such as veterinarians, oncologists, and pharmacologists be conducted to determine the best initial dosage and facilitate clinical trials on FZ in the near future.
10. Conclusions
Comprehensive verification through evidence-based medicine is crucial in reducing needless confusion in healthcare. Despite the potential anticancer capabilities of FZ, its pharmacokinetics, safety, and tolerance profiles in humans must be confirmed in extensive clinical trials before it can be used in any therapeutic context. Experts must further attempt to provide patients with reliable medical information.
Sultana T, Jan U, Lee H, Lee H, Lee JI. Exceptional Repositioning of Dog Dewormer: Fenbendazole Fever. Curr Issues Mol Biol. 2022 Oct 17;44(10):4977-4986. doi: 10.3390/cimb44100338. PMID: 36286053; PMCID: PMC9600184.
In 2022 Park et al performed an in vitro analysis of the anti-cancer effects of fenbendazole against a type of colorectal cancer cell that is resistant to one of the most commonly used drugs to treat cancer called Fluorouracil (“5FU” or “FU”), which is often used in combination with other cancer drugs. They discovered that it induces dose and time-dependent anti-proliferative effects. The mode of action is similar to that of ivermectin: induction of cell cycle arrest and induction of apoptosis, amongst other mechanisms. They found that toxicity to normal cells is low at concentrations that are inhibitory to cancer cells.
Anti-cancer effects of fenbendazole on 5-fluorouracil-resistant colorectal cancer cells (2022)
Abstract
Benzimidazole anthelmintic agents have been recently repurposed to overcome cancers resistant to conventional therapies. To evaluate the anti-cancer effects of benzimidazole on resistant cells, various cell death pathways were investigated in 5-fluorouracil-resistant colorectal cancer cells. The viability of wild-type and 5-fluorouracil-resistant SNU-C5 colorectal cancer cells was assayed, followed by Western blotting. Flow cytometry assays for cell death and cell cycle was also performed to analyze the anti-cancer effects of benzimidazole. When compared with albendazole, fenbendazole showed higher susceptibility to 5-fluorouracil-resistant SNU-C5 cells and was used in subsequent experiments. Flow cytometry revealed that fenbendazole significantly induces apoptosis as well as cell cycle arrest at G2/M phase on both cells. When compared with wild-type SNU-C5 cells, 5-fluorouracil-resistant SNU-C5 cells showed reduced autophagy, increased ferroptosis and ferroptosis-augmented apoptosis, and less activation of caspase-8 and p53. These results suggest that fenbendazole may be a potential alternative treatment in 5-fluorouracil-resistant cancer cells, and the anticancer activity of fenbendazole does not require p53 in 5-fluorouracil-resistant SNU-C5 cells.
Keywords: Apoptosis, Colorectal cancer, Drug resistance, Fenbendazole, p53
Drug repositioning of approved therapies might extend their therapeutic potential against resistant cancer cells [1]. Benzimidazole anthelmintic agents are relatively non-toxic to normal cells [2-6] with a half-maximal inhibitory concentration (IC50) of 5 µM for less sensitivity in 461 cancer cells [7]. A recent report summarized the anti-tumourigenic activity of benzimidazole as follows [1]: 1) cell cycle arrest at G2/M phase with increased levels of cyclin B1, p21 and p27Kip1; 2) apoptosis with increased expression of caspase-3, poly (ADP-ribose) polymerase (PARP), and cytochrome-C; 3) autophagy with increased microtubule-associated protein 1A/1B-light chain 3 (LC3) and Beclin-1; and 4) altered cell viability or differentiation with increased p53, p21, p38, and c-Jun N-terminal kinase (JNK), and decreased extracellular signal-regulated protein kinase (ERK).
Self-administration of fenbendazole, a benzimidazole anthelmintic agent, by patients with cancer has been reported in social media [8,9]. Although anti-cancer effects of fenbendazole as an alternative or supplementary agent were recently reported in a case series of genitourinary malignancies [10], no definitive evidence of anti-cancer effects exists in human because of its toxicity and teratogenicity [8]. Nevertheless, the potential for drug repositioning of fenbendazole requires further investigation because of the much higher IC50 values in normal cells compared with cancer cells [5].
Taken together, SNU-C5/5-FUR cells exhibited approximately 10-fold higher IC50 values with fenbendazole compared with SNU-C5 cells, but still showed cytotoxicity at micromolar concentrations as previously reported [35]. Fenbendazole induces apoptosis as well as cell cycle arrest at G2/M phase via p53-p21 pathways in CRC cells. In case of cell death, fenbendazole induces primarily apoptotic cell death rather than autophagy, ferroptosis, and necroptosis. Although fenbendazole has anti-cancer effects on both 5-FU-sensitive and resistant CRC cells, the mechanism of action appears to be different. That is, fenbendazole promotes cell death by activating p53-mediated apoptosis in SNU-C5 cells, whereas by both enhancing p53-independent apoptosis and ferroptosis-augmented apoptosis in SNU-C5/5-FUR cells. Therefore, further studies are needed to identify sensitive and resistant cancer cell types to facilitate cell type-specific treatment with benzimidazole.
Full paper:
Park D, Lee JH, Yoon SP. Anti-cancer effects of fenbendazole on 5-fluorouracil-resistant colorectal cancer cells. Korean J Physiol Pharmacol. 2022 Sep 1;26(5):377-387. doi: 10.4196/kjpp.2022.26.5.377. PMID: 36039738; PMCID: PMC9437363.
Teratogenicity: “Teratogens are factors that can alter normal intrauterine development of fetal growth, anatomic structures, physical functioning, and postnatal development.”
Interesting how experimental mRNA gene therapies can be administered despite near zero risk of COVID-19 to the mother or child and disturbing rates of miscarriage & infertility etc, but not fenbendazole:
Anthelmintics as Potential Anti-Cancer Drugs? (2020)
ANTI-CANCER EFFECTS OF ANTHELMINTICS
Anti-cancer effects of fenbendazole, albendazole, and mebendazole have been known against cancer cell lines in vitro for a long time.1 Microtubule destabilization is suggested as a mechanism of action.2 Based on these findings, clinical trials have been done for cancer patients with albendazole or mebendazole.3 One Swedish clinical trial was initiated on August 11, 2018 and terminated on January 22, 2020 due to lack of effect after enrollment of 11 cancer patients.4 There is no definite evidence of anti-cancer effects in human patients so far. Clinical trial with fenbendazole is impossible, because it is not permitted for human use due to toxicities.
SAFETY ISSUES
Food and Drug Administration (FDA) and European Medicines Agency (EMA) prohibit fenbendazole for human use. Recommended dosage for animal is 5 mg/kg, which is more than 100 fold acceptable daily intake for human safety (40 micrograms per kilogram body weight). The decision was based on toxicity and teratogenicity studies conducted in Hoechst Research Laboratories (NADA 128-620).5 Actual toxicities, such as acute hepatitis, following self-administration of fenbendazole are reported.
Heo DS. Anthelmintics as Potential Anti-Cancer Drugs? J Korean Med Sci. 2020 Feb 17;35(6):e75. doi: 10.3346/jkms.2020.35.e75. PMID: 32056406; PMCID: PMC7025903.
As per reference 5 above, for no stated reason (eg after testing clinically or in vitro) they applied a 100-fold safety factor, a maximum of 0.4mg/kg despite a 2019 study finding that “no significant side effects were observed in humans in response to the administration of the major FEN metabolite oxfendazole even at 60 mg kg−1 for 14 d”
FREEDOM OF INFORMATION SUMMARY
Muscle tolerance for PANACUR/SAFE-GUARD (fenbendazole) Suspension 10% in cattle and goats
VI. HUMAN FOOD SAFETY:
A. Toxicity Studies
Toxicity and teratogenicity studies were presented in the original NADA 128-620 for fenbendazole in cattle and were conducted in Hoechst Research Laboratories in Frankfort, Germany, and in the United States. No new toxicity studies were conducted to support this Supplement to NADA 128-620.
B. Acceptable Daily Intake
An Acceptable Daily Intake (ADI) of 40 micrograms per kilogram body weight per day for residues of fenbendazole has been assigned on the basis of the toxicity studies referenced above. The ADI of 40 mcg/kg bw/day was calculated from the no effect level for a six month dog study using a 100-fold safety factor.
C. Safe Concentrations
The safe concentrations for total residues of fenbendazole in cattle have been revised using the procedure described in the Federal Register, Volume 59, page 27499, July 22, 1994. The original and revised safe concentrations are listed below.
Tissue Original Safe Concentrations Revised Safe Concentrations
muscle 5 ppm 8 ppm
liver 10 ppm 24 ppm
kidney 15 ppm 48 ppm
fat 20 ppm 48 ppm
D. Tolerance in Liver
The tolerance for residues of parent fenbendazole in cattle liver was not recalculated with the approval of this Supplement, and the tolerance in cattle and goat liver remains unchanged at 800 ppb.
E. Muscle Tolerance Assignment
A muscle tolerance of 400 ppb is assigned as the tolerance for residues of fenbendazole measured as parent fenbendazole in cattle muscle. The residue studies conducted with 14C-fenbendazole for the original approval in cattle showed that total residues in muscle tissue are well below the safe concentration of 8 ppm even at short withdrawal times following the approved conditions of use of the drug. The muscle tolerance value of 400 ppb is in the range of the maximum fenbendazole values expected in cattle muscle at one to two days post dosing.
Residues of parent fenbendazole are not detectable by the official regulatory assay at the regulated withdrawal time of eight days for the paste and suspension formulations. The tolerance of 400 ppb will also serve as the tolerance in muscle tissue of goats, which are a minor species approved under NADA 128-620. Residues of fenbendazole below 400 ppb in cattle or goat muscle indicate that total residues in muscle are below the safe concentration. However, residues below that level in muscle are not indicative of the safety of residues in other edible tissues.
In various studies with rodents this appears to be a case of caution or confliction not borne out from any experimental findings. Its used at normal doses in many pregnant animals:
The behavioral teratogenic potential of fenbendazole: a medication for pinworm infestation (2000)
Abstract
Fenbendazole (FBZ) is a benzimidazole currently used for anthelmintic treatment of pinworm populations in numerous animal species although it is not currently approved for laboratory rodents in the U.S. It has received considerable interest for treating rodent populations due to its low toxicity, wide safety margin and apparent absence of gross teratogenic effects. The purpose of this study was to assess the behavioral teratogenic potential of FBZ. Pregnant rats were administered either FBZ-medicated feed at a therapeutic level or normal rat chow throughout pregnancy and gestation. FBZ had no effect on pregnancy indicators such as maternal weight gain or water consumption, number of pups born or pup birth weights. Offspring were examined in a variety of paradigms including righting reflex, negative geotaxis, running wheel activity, Morris water maze (MWM) performance and digging maze performance. FBZ offspring did show delayed righting reflex, some modest changes in locomotor activity in a running wheel and minor alterations in performance during the probe session of the MWM relative to controls. However, the effects of FBZ on behavior were subtle and many of the behaviors examined were unaffected. These results suggest that FBZ may be an effective and relatively safe anthelmintic treatment for use in breeding colonies.
Barron S, Baseheart BJ, Segar TM, Deveraux T, Willford JA. The behavioral teratogenic potential of fenbendazole: a medication for pinworm infestation. Neurotoxicol Teratol. 2000 Nov-Dec;22(6):871-7. doi: 10.1016/s0892-0362(00)00102-1. PMID: 11120393.
A more recent study didn’t find any evidence of humanteratogenic or carcinogenic risks:
Risk Assessment of Human Consumption of Meat From Fenbendazole-Treated Pheasants (2021)
Abstract
Fenbendazole is a benzimidazole-class anthelmintic that is used for the control of immature and adult stages of internal parasites, such as nematodes and trematodes, in domestic food-animal species. It is not approved by the United States Food and Drug Administration for treating pheasants despite Syngamus trachea being one of the most prevalent nematodes that parasitize pheasants. Because it is a highly effective treatment, e.g., 90% effectiveness against S. trachea, and there are very few alternative therapeutic options, this anthelminthic is used in an extra-label manner in the pheasant industry, but few studies have been conducted assessing risks to humans. Therefore, we conducted a risk assessment to evaluate the potential repeat-dose and reproductive, teratogenic, and carcinogenic human risks that may be associated with the consumption of tissues from pheasants that were previously treated with fenbendazole. We conducted a quantitative risk assessment applying both deterministic and stochastic approaches using different fenbendazole sulfone residue limits (tolerance, maximum residue limits, and analytical limit of detection) established in different poultry species by the Food and Drug Administration, the European Medicines Agency, and other regulatory agencies in Japan, Turkey, and New Zealand. Our results show that fenbendazole poses minimal risk to humans when administered to pheasants in an extra-label manner, and a comparison of different fenbendazole sulfone residue limits can help assess how conservative the withdrawal interval should be after extra-label drug use.
Keywords: food safety, risk analysis, drug residue, stochastic model, poultry, extra-label use
Carreño Gútiez M, Tell LA, Martínez-López B. Risk Assessment of Human Consumption of Meat From Fenbendazole-Treated Pheasants. Front Vet Sci. 2021 Jun 4;8:665357. doi: 10.3389/fvets.2021.665357. PMID: 34150886; PMCID: PMC8212976.
With experimental gene therapies none of this concern applies. At best there is no long term safety data available being referred to here:
Pregnancy, breastfeeding, fertility and coronavirus (COVID-19) vaccination
It'sstrongly recommended that you get vaccinated against coronavirus (COVID-19) if you're pregnant or breastfeeding.
If you're pregnant
If you're pregnant, it's important to get vaccinated to protect you and your baby. The antibodies your body produces in response to the vaccine can also give your baby protection against COVID-19.
You're at higher risk of getting seriously ill from COVID-19 if you're pregnant. If you get COVID-19 late in your pregnancy, your baby could also be at risk.
Evidence shows that most pregnant women with COVID-19 who need hospital treatment or intensive care in the UK have not been vaccinated.
If you're pregnant and have not had your first 2 doses and booster dose yet, it's important to get your vaccinations as soon as possible.
It's safe to have the vaccine during any stage of pregnancy, from the first few weeks up to your expected due date. You do not need to delay vaccination until after you have given birth.
Getting vaccinated against COVID-19 reduces the risk of having a stillbirth.
There's no evidence COVID-19 vaccination increases the risk of having a miscarriage, pre-term birth or other complications in your pregnancy.
The COVID-19 vaccines do not contain any live viruses and cannot give you or your baby COVID-19.
They have been widely used during pregnancy in other countries and there have been no safety concerns. In the UK, over 100,000 pregnant women have been vaccinated.
And yet caution reigns supreme with anything else. Total regulatory capture:
Foods to avoid in pregnancy
What you can eat
pasteurised or unpasteurised hard cheeses, such as cheddar, Gruyere and parmesan
pasteurised semi-hard cheeses, such as Edam and Stilton
pasteurised soft cheeses, such as cottage cheese, mozzarella, feta, cream cheese, paneer, ricotta, halloumi, goats' cheese without a white coating on the outside (rind) and processed cheese spreads
soft or blue cheese (pasteurised or unpasteurised) that has been cooked until steaming hot
pasteurised milk, yoghurt, cream and ice cream
What to avoid
any other foods made from unpasteurised milk, such as soft ripened goats' cheese
pasteurised or unpasteurised mould-ripened soft cheeses with a white coating on the outside, such as Brie, Camembert and chèvre (unless cooked until steaming hot)
pasteurised or unpasteurised soft blue cheeses, such as Danish blue, Gorgonzola and Roquefort (unless cooked until steaming hot)
unpasteurised cows' milk, goats' milk, sheep's milk or cream
Why
There's a small chance that unpasteurised or soft ripened dairy products may contain Listeria bacteria. This can cause an infection called listeriosis.
Listeriosis can lead to miscarriage or stillbirth, or make your newborn baby very unwell.
Soft cheeses with a white coating on the outside have more moisture. This can make it easier for bacteria to grow.
Cooking cheese until it's steaming hot kills bacteria, reducing the risk of listeriosis.
Returning to fenbendazole, other studies of interest.
This one’s great as they didn’t go looking for it, they were just using it as a pinworm treatment with low adverse effects and the coincident lymphoma xenografts failed to grow:
Unexpected Antitumorigenic Effect of Fenbendazole when Combined with Supplementary Vitamins (2008)
Abstract
Diet containing the anthelminthic fenbendazole is used often to treat rodent pinworm infections because it is easy to use and has few reported adverse effects on research. However, during fenbendazole treatment at our institution, an established human lymphoma xenograft model in C.B-17/Icr-prkdcscid/Crl (SCID) mice failed to grow. Further investigation revealed that the fenbendazole had been incorporated into a sterilizable diet supplemented with additional vitamins to compensate for loss during autoclaving, but the diet had not been autoclaved. To assess the role of fenbendazole and supplementary vitamins on tumor suppression, 20 vendor-supplied 4-wk-old SCID mice were assigned to 4 treatment groups: standard diet, diet plus fenbendazole, diet plus vitamins, and diet plus both vitamins and fenbendazole. Diet treatment was initiated 2 wk before subcutaneous flank implantation with 3 × 107 lymphoma cells. Tumor size was measured by caliper at 4-d intervals until the largest tumors reached a calculated volume of 1500 mm3. Neither diet supplemented with vitamins alone nor fenbendazole alone caused altered tumor growth as compared with that of controls. However, the group supplemented with both vitamins and fenbendazole exhibited significant inhibition of tumor growth. The mechanism for this synergy is unknown and deserves further investigation. Fenbendazole should be used with caution during tumor studies because it may interact with other treatments and confound research results.
Abbreviation: HIF, hypoxia-inducible factor 1α
Gao P, Dang CV, Watson J. Unexpected antitumorigenic effect of fenbendazole when combined with supplementary vitamins. J Am Assoc Lab Anim Sci. 2008 Nov;47(6):37-40. PMID: 19049251; PMCID: PMC2687140.
Further experiments in mice could not directly reproduce this effect, but the researchers concluded that this was because the type of tumor, mammary, was less sensitive to microtubule inhibitors then the human lymphoma xenograft model used in the previous study - leukaemia cells appear easier to treat with tubulin inhibitors, but it varies by solid tumor microenvironment too.
By “fenbendazole could have significant antitumor effects when given insufficiently intensive regimens” they mean maximising the dose and/or administering co-treatments:
Use of fenbendazole-containing therapeutic diets for mice in experimental cancer therapy studies (2012)
We examined the effect of feeding a therapeutic diet containing 150 ppm fenbendazole on the growth of EMT6 mouse mammary tumors implanted into BALB/c Rw mice. Mice were randomized to receive either a fenbendazole-containing or control diet for 1 wk before tumor cells were injected intradermally in the flanks and throughout tumor growth. Tumor growth was monitored by serial measurements of tumor diameters from the time tumors became palpable until they reached 1000 mm3. The medicated diet did not alter tumor growth, invasion, or metastasis. When tumors reached volumes of approximately 100 mm3, some were irradiated locally with 10 Gy of X-rays. Irradiation significantly delayed tumor growth; fenbendazole did not alter the radiation-induced growth delay. However, cell culture studies showed that fenbendazole concentrations not far above those expected in the tissues of mice on this diet altered the growth of the tumor cells in culture. Recent data from other laboratories also have demonstrated effects of fenbendazole that could complicate experiments. Care should therefore be exercised in deciding whether chow containing fenbendazole should be administered to mouse colonies being used in cancer research.
A fenbendazole-containing diet was reported to inhibit the growth of a human lymphoma xenograft in SCID mice and to produce significant increases in total WBC and neutrophil counts, but only when given with high-dose vitamins. 9It is unclear whether the changes in the growth of these xenografts reflected a direct effect of the drug on the tumor cells or stimulation of an immune response that inhibited the growth of the human tumors, which can survive xenotransplantation only in severely immunodeficient hosts. Other data suggest that fenbendazole can act as an antitumor agent. A presentation at the 2010 meeting of the American Association of Cancer Research3 reported that high doses of fenbendazole, albendazole, and mebendazole inhibited the growth of paclitaxel-resistant tumors. Because fenbendazole acts by inhibiting microtubule formation5,16 and several widely used anticancer drugs produce their antineoplastic effects by disrupting either microtubule formation (vincristine, vinblastine)36 or microtubule depolymerization (paclitaxel, docetaxel),36fenbendazole could have significant antitumor effects when given in sufficiently intensive regimens.
It is therefore possible that tumor cell lines that are more sensitive to the cytotoxic effects of either tubulin-directed agents or the metabolic disruption induced by benzimidazoles might be more sensitive to fenbendazole than are EMT6 tumors. In this regard, it is notable that experiments demonstrating that fenbendazole inhibited the growth of a human leukemia in SCID mice9 used a model system with notable sensitivity in 2 respects. First, leukemia cells (like most cells of hematopoietic origin) are less able to repair damage and more likely to undergo apoptosis in response to injury from radiation, drugs or metabolic disruption than are cells of other lineages.11,36 Second, the cells in the cited study9 were xenografted into SCID mice, which are used as hosts for xenografts because they have an mutation that produces defective V(D)J joining, producing deficits in T and B cell immunity that prevent the mice from rejecting grafts of foreign tissues.1 However, this same mutation also prevents the repair of DNA double-strand breaks produced by radiation, anticancer drugs, and other agents and increases the radiation sensitivity of the bone marrow cells in SCID mice by a factor of 3 over that of the cells in normal BALB/c mice.1 Both the tumors and the hosts used in the earlier studies9 therefore would be expected to be unusually sensitive to the effects of agents that damaged DNA directly or indirectly through effects on metabolic processes or tubulin.
The effects of fenbendazole on tubulin might synergize with or antagonize the effects of anticancer drugs with mechanisms of action that involve stabilization or disruption of microtubules (for example, paclitaxel, docetaxel, vincristine, vinblastine, colchicine and podophyllotoxin);36 such synergism has been reported for the closely related benzimidazole flubendazole.35 The possibility of such interactions would not be anticipated by the many cancer researchers who are unaware that the mechanism of action of this antihelminth involves altered tubulin stability. Furthermore, it is possible that fenbendazole would have effects that varied within the different microenvironments within solid tumors. If this effect occurred, the interactions of fenbendazole with anticancer drugs having activity that is influenced by these tumor microenvironments25-27 could be complex. The effects of the benzimidazoles on glucose uptake and metabolism6,15,16,37 raise the possibility of interactions between fenbendazole and drugs that are activated in hypoxia or drugs that alter energy metabolism. Therefore, significant interactions between fenbendazole and anticancer drugs having several different mechanisms of action are quite possible.
Characteristics of the mice also might influence the effects of fenbendazole on experiments. Mice that have greater drug absorption from the intestine because of hereditary disease phenotypes, certain microbiologic profiles (induced by intercurrent or experimental infections), or concomitant treatment with drugs that alter absorption might take up more of this poorly absorbed drug and therefore have higher blood and tissue levels of fenbendazole and its active metabolites,15,18,21 which in turn could produce greater confounding effects. Tumor–host systems that are especially sensitive to small changes in the host immune system, such as tumor xenografts in immunocompromised animals or tumors that are immunogenic in the rodent strain of origin,31 might also be more sensitive to the subtle immunologic effects of fenbendazole.
Because of these potential problems, we advise caution when fenbendazole-containing diets are used to treat rodent colonies actively used in research on experimental cancer therapy.
Duan Q, Liu Y, Booth CJ, Rockwell S. Use of fenbendazole-containing therapeutic diets for mice in experimental cancer therapy studies. J Am Assoc Lab Anim Sci. 2012 Mar;51(2):224-30. PMID: 22776123; PMCID: PMC3314526.
Fenbendazole Suppresses Growth and Induces Apoptosis of Actively Growing H4IIE Hepatocellular Carcinoma Cells via p21-Mediated Cell-Cycle Arrest (2022)
Abstract
Bendimidazole anthelmintics (BAs) have gained interest for their anticancer activity. The anticancer activity is mediated via multiple intracellular changes, which are not consistent under different conditions even in the same cells. We investigated the anticancer activity of fenbendazole (FZ, one of BAs) under two different growth conditions. The growth rate of H4IIE cells was dose-dependently decreased by FZ only in actively growing cells but not in fully confluent quiescent cells. Apoptosis-associated changes were also induced by FZ in actively growing cells. Markers of autophagy were not changed by FZ. The number of cells was markedly increased in sub-G1 phase but decreased in S- and G2/M phases by FZ. FZ up-regulated p21 (an inhibitor of cyclin-CDK) but suppressed the expression of cell cycle-promoting proteins (cyclin D1 and cyclin B1). FZ did not affect integrin αV or n-cadherin expression as well as cell migration. Glycolytic changes (glucose consumption and lactate production) and the generation of reactive oxygen species (ROS) were not affected by FZ. Although the activity of mitogen-activated protein kinases (MAPKs) was altered by FZ, the inhibition of MAPKs did not affect the pro-apoptotic activity of FZ. Taken together, FZ selectively suppressed the growth of cells via p21-mediated cell cycle arrest at G1/S and G2/M, and resulted in apoptosis only in actively growing cells but not in quiescent cells. Glucose metabolism, ROS generation, and MAPKs are unlikely targets of FZ at least in H4IIE rat hepatocellular carcinoma cells used in this study.
Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways (2018)
Abstract
Drugs that are already clinically approved or experimentally tested for conditions other than cancer, but are found to possess previously unrecognized cytotoxicity towards malignant cells, may serve as fitting anti-cancer candidates. Methyl N-(6-phenylsulfanyl-1H benzimidazol-2-yl) carbamate [Fenbendazole, FZ], a benzimidazole compound, is a safe and inexpensive anthelmintic drug possessing an efficient anti-proliferative activity. In our earlier work, we reported a potent growth-inhibitory activity of FZ caused partially by impairment of proteasomal function. Here, we show that FZ demonstrates moderate affinity for mammalian tubulin and exerts cytotoxicity to human cancer cells at micromolar concentrations. Simultaneously, it caused mitochondrial translocation of p53 and effectively inhibited glucose uptake, expression of GLUT transporters as well as hexokinase (HK II) - a key glycolytic enzyme that most cancer cells thrive on. It blocked the growth of human xenografts in nu/nu mice model when mice were fed with the drug orally. The results, in conjunction with our earlier data, suggest that FZ is a new microtubule interfering agent that displays anti-neoplastic activity and may be evaluated as a potential therapeutic agent because of its effect on multiple cellular pathways leading to effective elimination of cancer cells.
Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. 2018 Aug 9;8(1):11926. doi: 10.1038/s41598-018-30158-6. PMID: 30093705; PMCID: PMC6085345.
Fenbendazole and its synthetic analog interfere with HeLa cells' proliferation and energy metabolism via inducing oxidative stress and modulating MEK3/6-p38-MAPK pathway (2022)
Abstract
Fenbendazole, a broad-spectrum anti-parasitic drug, can be a potential anti-tumor agent. In this study, we synthesized and purified its derivative, analog 6, intending to achieve improved efficacy in cancer cells and decreased toxicity in normal cells. To evaluate in vitro anti-tumor activities of fenbendazole and analog 6 in different cancer cell lines, a CCK-8 assay was performed, and we found that human cervical cancer HeLa cells were more sensitive to analog 6 than to fenbendazole. Furthermore, we explored the associated mechanism, and our results showed that analog 6 and fenbendazole could induce oxidative stress by accumulating ROS. It not only activated the p38-MAPK signaling pathway, thereby inhibiting the proliferation of HeLa cells and enhancing the apoptosis of HeLa cells, but also significantly induced impaired energy metabolism and restrained their migration and invasion. In addition, the modified analog 6 showed reduced toxicity to normal cells without decreased anti-cancer effect. In conclusion, fenbendazole and analog 6 have multiple targets and strong anti-tumor effects on HeLa cells in vitro and in vivo. The optimized analog 6 could inhibit the viability of HeLa cells with lower toxicity than normal human cells, promising to be developed as an antitumor active compound.
Keywords: Analogs; Antitumor activity; Fenbendazole; Human cervical cancer; Oxidative stress.
Peng Y, Pan J, Ou F, Wang W, Hu H, Chen L, Zeng S, Zeng K, Yu L. Fenbendazole and its synthetic analog interfere with HeLa cells' proliferation and energy metabolism via inducing oxidative stress and modulating MEK3/6-p38-MAPK pathway. Chem Biol Interact. 2022 Jul 1;361:109983. doi: 10.1016/j.cbi.2022.109983. Epub 2022 May 13. PMID: 35569513.
Antiviral effects of fenbendazole, another factor it has in common with ivermectin to some degree. Useful properties for treating co-infections too:
Dewormer drug fenbendazole has antiviral effects on BoHV-1 productive infection in cell cultures (2020)
Abstract
Background
Fenbendazole, a dewormer drug, is used widely in the clinical treatment of parasite infections in animals. Recent studies have shown that fenbendazole has substantial effects on tumor growth, immune responses, and inflammatory responses, suggesting that fenbendazole is a pluripotent drug. Nevertheless, the antiviral effects have not been reported. Fenbendazole can disrupt microtubules, which are essential for multiple viruses infections, suggesting that fenbendazole might have antiviral effects.
Objectives
This study examined whether fenbendazole could inhibit bovine herpesvirus 1 (BoHV-1) productive infection in cell cultures.
Methods
The effects of fenbendazole on viral production, transcription of the immediate early (IE) genes, viron-associated protein expression, and the cellular signaling PLC-γ1/Akt pathway were assessed using distinct methods.
Results
Fenbendazole could inhibit BoHV-1 productive infections significantly in MDBK cells in a dose-dependent manner. A time-of-addition assay indicated that fenbendazole affected both the early and late stages in the virus replication cycles. The transcription of IE genes, including BoHV-1 infected cell protein 0 (bICP0), bICP4, and bICP22, as well as the synthesis of viron-associated proteins, were disrupted differentially by the fenbendazole treatment. The treatment did not affect the cellular signaling pathway of PLC-γ1/Akt, a known cascade playing important roles in virus infection.
Conclusions
Overall, fenbendazole has antiviral effects on BoHV-1 replication.
Keywords: Bovine herpesvirus 1, immediate early gene, fenbendazole, Akt
Chang L, Zhu L. Dewormer drug fenbendazole has antiviral effects on BoHV-1 productive infection in cell cultures. J Vet Sci. 2020 Sep;21(5):e72. doi: 10.4142/jvs.2020.21.e72. PMID: 33016019; PMCID: PMC7533386.
This study was an attempt to improve bioavailability, but provides useful safety and efficacy data:
Physicochemical, Pharmacokinetic, and Toxicity Evaluation of Soluplus® Polymeric Micelles Encapsulating Fenbendazole (2020)
Abstract
Fenbendazole (FEN), a broad-spectrum benzimidazole anthelmintic, suppresses cancer cell growth through various mechanisms but has low solubility and achieves low blood concentrations, which leads to low bioavailability. Solubilizing agents are required to prepare poorly soluble drugs for injections; however, these are toxic. To overcome this problem, we designed and fabricated low-toxicity Soluplus® polymeric micelles encapsulating FEN and conducted toxicity assays in vitro and in vivo. FEN-loaded Soluplus® micelles had an average particle size of 68.3 ± 0.6 nm, a zeta potential of −2.3 ± 0.2 mV, a drug loading of 0.8 ± 0.03%, and an encapsulation efficiency of 85.3 ± 2.9%. MTT and clonogenic assays were performed on A549 cells treated with free FEN and FEN-loaded Soluplus® micelles. The in vitro drug release profile showed that the micelles released FEN more gradually than the solution. Pharmacokinetic studies revealed lower total clearance and volume of distribution and higher area under the curve and plasma concentration at time zero of FEN-loaded Soluplus® micelles than of the FEN solution. The in vivo toxicity assay revealed that FEN-loaded Soluplus® micelle induced no severe toxicity. Therefore, we propose that preclinical and clinical safety and efficacy trials on FEN-loaded Soluplus® micelles would be worthwhile.
Certain cancers have high mortality rates in humans. Therefore, the development of safe anticancer drugs with few side effects is an important research objective in pharmaceutical science. Fenbendazole (FEN) is a broad-spectrum benzimidazole anthelmintic. It has been widely used in veterinary medicine and induces no significant side effects.The European Medicines Agency has not established a no observable adverse effects level (NOAEL) for single-dose FEN administration but recommended a NOAEL of 4 mg kg−1 BW d−1 for repeated FEN administration. Moreover, no significant side effects were observed in humans in response to the administration of the major FEN metabolite oxfendazole even at 60 mg kg−1 for 14 d [1]. Benzimidazole (BZD) anthelmintic agents have been studied in recent years for their anticancer effects [2,3]. Several papers have reported their modes of action and positive effects [4,5]. FEN suppresses cancer cell growth through various mechanisms. It inhibits proteasomal activity and induces endoplasmic reticulum stress and reactive oxygen species-dependent apoptosis [6]. Second, it has anti-tubulin efficacy [5]. Other BZDs arrest mitosis and promote apoptosis. Mebendazole displays anti-tubulin and antitumor efficacy in vivo [7]. However, FEN has low solubility and achieves only low blood concentrations [8,9]. The limited solubility makes it difficult to develop parenteral and even topical preparations. For these reasons, it has a low area under the curve (AUC) and poor bioavailability
Figure 5. In vitro clonogenic assay of free fenbendazole (FEN)- and FEN-loaded Soluplus® micelle-treated A549 cell line, ** p < 0.01. https://www.mdpi.com/1999-4923/12/10/1000#
Figure 8. Total amount of fenbendazole (FEN) in each organ at 1 and 8 h after administration of FEN in 25% Cremophor EL®/EtOH solution and FEN-loaded Soluplus® micelles, ** p < 0.01, *** p < 0.001. https://www.mdpi.com/1999-4923/12/10/1000#
The IC50 for free FEN drug was 3070 nM (3.07 μM). Curcumin has an IC50 range of 5.43–108.69 μM, and its anticancer action is downregulation of the BCL-2 family. 17-AAG has an IC50 range of 0.1–2.37 μM, and its anticancer mechanism is the suppression of heat shock protein 90. Hence, the IC50 of FEN indicates that the drug has sufficient anticancer efficacy [43,44,45]. The clonogenic assay confirmed the long-term inhibition of cell reproduction. It revealed that the FEN micellar formulation entirely inhibited colony formation at 1740 μM but was also effective at only 174 μM.
Jin IS, Jo MJ, Park C-W, Chung YB, Kim J-S, Shin DH. Physicochemical, Pharmacokinetic, and Toxicity Evaluation of Soluplus® Polymeric Micelles Encapsulating Fenbendazole.Pharmaceutics. 2020; 12(10):1000. https://doi.org/10.3390/pharmaceutics12101000
Unbiased Phenotype-Based Screen Identifies Therapeutic Agents Selective for Metastatic Prostate Cancer (2021)
Abstract
In American men, prostate cancer is the second leading cause of cancer-related death. Dissemination of prostate cancer cells to distant organs significantly worsens patients' prognosis, and currently there are no effective treatment options that can cure advanced-stage prostate cancer. In an effort to identify compounds selective for metastatic prostate cancer cells over benign prostate cancer cells or normal prostate epithelial cells, we applied a phenotype-based in vitro drug screening method utilizing multiple prostate cancer cell lines to test 1,120 different compounds from a commercial drug library. Top drug candidates were then examined in multiple mouse xenograft models including subcutaneous tumor growth, experimental lung metastasis, and experimental bone metastasis assays. A subset of compounds including fenbendazole, fluspirilene, clofazimine, niclosamide, and suloctidil showed preferential cytotoxicity and apoptosis towards metastatic prostate cancer cells in vitro and in vivo. The bioavailability of the most discerning agents, especially fenbendazole and albendazole, was improved by formulating as micelles or nanoparticles. The enhanced forms of fenbendazole and albendazole significantly prolonged survival in mice bearing metastases, and albendazole-treated mice displayed significantly longer median survival times than paclitaxel-treated mice. Importantly, these drugs effectively targeted taxane-resistant tumors and bone metastases - two common clinical conditions in patients with aggressive prostate cancer. In summary, we find that metastatic prostate tumor cells differ from benign prostate tumor cells in their sensitivity to certain drug classes. Taken together, our results strongly suggest that albendazole, an anthelmintic medication, may represent a potential adjuvant or neoadjuvant to standard therapy in the treatment of disseminated prostate cancer.
A discussion piece for licensed medical practitioners only. Not an endorsement or promotion. Not checked for efficacy, safety or interactions:
Fenbendazole Cancer Protocol
Step By Step Guide
The Fenbendazole Cancer Protocol has been gaining rapid interest over the past year following some fenbendazole cancer success stories. The most recognized one is of a man diagnosed with stage 4 small cell Lung cancer that sent home with a 3-month life expectancy. He started taking fenbendazole (Panacur C), a dog dewormer, with some additional supplements. The following PET scan showed remarkable improvement, and after a few months, he was declared cancer-free.
Fenbendazole Protocol – A Simple Step-by-Step Guide
The basic fenbendazole protocol people follow is surprisingly simple and includes a few added supplements to the fenbendazole:
Fenbendazole which has 222mg of Fenbendazole per gram: one packet of powder per day for seven days a week. It can be mixed with food such as yogurt or simply taken by itself.
It is advised to purchase Panacur C or Safeguard brands only, as both are regulated and have been consistent in third-party lab results.
2. Curcumin: 600 mg a Day
600 mg per day of bioavailable curcumin, which is the active agent in the herb turmeric. Curcumin may help increase healthy p53 levels, and it has been shown to be a potentially effective cancer therapy supplement.
3. CBD Oil: 25 mg a Day
25 milligrams daily, taken sublingually (under the tongue). The CBD oil should be high-purity level broad-spectrum. CBD has been shown to potentially modulate tumor growth
To enhance CBD healing response for cancer symptoms, slowly increase to .5ml of CBD twice daily for a total of 50 mg.
Some people using this protocol use CBD oil, while others choose to add THC. If you are considering adding THC, it is advised to use a Medical Cannabis Professional. Click here to speak with a nurse.
4. Berberine: 2-3 times a day
Berberine has shown important anti-tumor effects in numerous studies. These studies reported that Berberine could prevent the multiplication of cancer cells and inhibit metastasis and the spread of cancer cells. Berberine can work with Fenbendazole to further limit the cancer cells’ ability to take up glucose. This way, cancer cells are weakened and starved.
5. Quercetin: 1-2 a day
Due to its antioxidant, anti-tumor, and anti-inflammatory activity, quercetin has been studied extensively. Quercetin can inhibit the spread of many cancers such as prostate, cervical, lung, breast, and colon. Quercetin is not harmful to healthy cells yet powerful against cancer cells, making it a good candidate for a supplementary factor along with other anticancer medications.
Fenbendazole for Humans – Side Effects
Some research suggests that those who are weak from chemotherapy may experience more side effects than those not receiving conventional cancer treatment.
Some common side effects that have been reported include elevated liver enzymes, mild diarrhea, and mild stomach discomfort.
If you are currently taking chemotherapy for your cancer, it is best to discuss how to add curcumin and vitamin E with a medical professional.
Fenbendazole Cancer Protocol: The Scientific Data
As surprising as it sounds, there is documented research about deworming medications and their effect on cancer. This dry and tasteless Fenbendazole powder has been shown to exhibit “significant inhibition of tumor growth” when supplemented with vitamins A, D, E, K, and B.
Fenbendazole is a triple-threat to cancer: it kills cancer cells in three ways that are significant:
It destroys microtubules that sustain the structure of the cancer cell and its ability to divide and multiply rapidly.
It interrupts the cancer cells’ ability to process sugar, and cancer cells must metabolize sugar to survive.
It boosts the production of a cancer-killing gene called p53; a gene cancer patients may lack. When p53 becomes mutated or can’t keep cancer cells in check, cancer cells can proliferate.
The dewormer also works against parasites, which might be the origin of some cancers.
Other Research That Supports the Use of De-Wormers for Cancer
There’s another “sister” drug of Fenbendazole, called Mebendazole, a de-wormer medication prescribed to treat parasitic worm infections in humans. Mebendazole has shown promising results in treating cancer (Lung, Melanoma, Glioblastoma, Colon, and others).
Cases of aggressive brain cancers, including of younger patients, are increasing markedly. I would consider it a particularly nasty cancer.
Part of the reason for the case counts may well be the oncogenic effects of cleaved Spike S1 with gp120 inserts. It easily crosses the blood-brain barrier and accumulates:
…Glioma cells treated with gp120 (100 ng/mL for 7–10 days) showed higher proliferation rates and upregulation in the expression of enolase 2, hexokinase and glyceraldehyde-3-phosphate dehydrogenase when compared to untreated cells. Furthermore, we detected an increase in the activity of pyruvate kinase and a higher glycolytic index in gp120 treated cells. Gp120 treated GBM cells also showed heightened lipid and protein synthesis. Overall, we demonstrate that in glioma cells, the HIV envelope glycoprotein promotes proliferation and activation of glycolysis resulting in increased protein and lipid synthesis.
From: “HIV-1 Envelope Protein gp120 Promotes Proliferation and the Activation of Glycolysis in Glioma Cell”, (2018)
…Our results revealed the accumulation of the spike protein in the skull marrow, brain meninges, and brain parenchyma. The injection of the spike protein alone caused cell death in the brain, highlighting a direct effect on brain tissue. Furthermore, we observed the presence of spike protein in the skull of deceased long after their COVID-19 infection, suggesting that the spike’s persistence may contribute to long-term neurological symptoms. The spike protein was associated with neutrophil-related pathways and dysregulation of the proteins involved in the PI3K-AKT as well as complement and coagulation pathway. Overall, our findings suggest that SARS-CoV-2 spike protein trafficking from CNS borders into the brain parenchyma and identified differentially regulated pathways may present insights into mechanisms underlying immediate and long-term consequences of SARS-CoV-2 and present diagnostic and therapeutic opportunities.
From: “SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-Brain Axis: Potential Implications for Long-Term Neurological Complications in post-COVID-19” (2023)
Glioblastoma is the most lethal primary central nervous system cancer. Part of the reason the tumor is so deadly is because it is hard to treat. The tumor itself is invasive and aggressive – it develops tentacles that invade other areas. This makes it hard to completely remove with surgery. Further, the cancerous cells can grow and spread quickly in the brain.
Chemotherapy, radiation, and surgery have been considered treatment staples for glioblastoma for decades. However, many chemotherapy drugs are not able to reach the brain. There are also no specific treatments that can kill all the cancerous cells. Because of this, the tumor usually grows back within six to nine months of initial diagnosis and treatment. Notably, glioblastoma cells can also survive treatment by changing or adapting to their environment. Being able to predict how these changes occur is a crucial step to increasing patient survival.
From: “New Study Explores How Glioblastoma Cells Can Undergo Exploratory Adaptation to Survive and Cause Recurrence” (2020)
Benzimidazoles, including fenbendazole, may at least hold it in check for while and improve the quality of life or reduce the risk of recurrences.
There are few other options available.
Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle, (2021)
Abstract
Glioblastoma multiforme (GBM) is the most malignant and lethal primary brain tumor in adults accounting for about 50% of all gliomas. The only treatment available for GBM is the drug temozolomide, which unfortunately has frequent drug resistance issue. By analyzing the hub genes of GBM via weighted gene co-expression network analysis (WGCNA) of the cancer genome atlas (TCGA) dataset, and using the connectivity map (CMAP) platform for drug repurposing, we found that multiple azole compounds had potential anti-GBM activity. When their anti-GBM activity was examined, however, only three benzimidazole compounds, i.e. flubendazole, mebendazole and fenbendazole, potently and dose-dependently inhibited proliferation of U87 and U251 cells with IC50 values below 0.26 μM. Benzimidazoles (0.125−0.5 μM) dose-dependently suppressed DNA synthesis, cell migration and invasion, and regulated the expression of key epithelial-mesenchymal transition (EMT) markers in U87 and U251 cells. Benzimidazoles treatment also dose-dependently induced the GBM cell cycle arrest at the G2/M phase via the P53/P21/cyclin B1 pathway. Furthermore, the drugs triggered pyroptosis of GBM cells through the NF-κB/NLRP3/GSDMD pathway, and might also concurrently induced mitochondria-dependent apoptosis. In a nude mouse U87 cell xenograft model, administration of flubendazole (12.5, 25, and 50 mg · kg−1 · d−1, i.p, for 3 weeks) dose-dependently suppressed the tumor growth without obvious adverse effects. Taken together, our results demonstrated that benzimidazoles might be promising candidates for the treatment of GBM.
Keywords: glioblastoma, benzimidazoles, cell cycle arrest, apoptosis, pyroptosis, drug repurposing
Interestingly, we found that only benzimidazole compounds had anti-GBM activity, while imidazoles and triazoles did not. Flubendazole, fenbendazole and mebendazole all demonstrated good time- and dose-dependent anti-GBM activity. Mebendazole had been reported to potentiate radiation therapy in triple-negative breast cancer [31]. Mebendazole inhibited tumor growth and prevented lung metastasis in models of advanced thyroid cancer [32] and resulted in increasing survival in two preclinical models of GBM [33]. Fenbendazole was identified as a microtubule-destabilizing agent and caused human NSCLC cell death [34]. Flubendazole elicited anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in triple-negative breast cancer [35, 36]. It also exerted potent antitumor activity in colorectal cancer cells by inhibiting STAT3 and activating autophagy [37].
From: “Fig. 9 Schematic model for the mechanism of action of benzimidazoles. Benzimidazoles induced concurrent apoptotic and pyroptotic cell death of glioblastoma cells by arresting the cell cycle”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8724275/
In conclusion, benzimidazoles are promising candidates for the treatment of GBM via arresting cell cycle and inducing concurrent apoptotic and pyroptotic cell death. It is of great importance to develop benzimidazoles for combination therapy or as alternative drugs for GBM patients who are resistant or less responsive to TMZ.
We only have animal studies to refer to from trials investigating parasitology, but interesting nonetheless. They do co-administer them for animal use:
Fenbendazole / Ivermectin is a combination compound of an anthelmintic and anti-parasitic medications. Fenbendazole / Ivermectin is commonly prescribed for treatment and prevention of worms and parasites.
Efficacy of fenbendazole and ivermectin in treating gastrointestinal nematode infections in an Ontario cow-calf herd (2019)
It would have been beneficial to have the treatments occur later in the grazing period, when the infection pressure could be determined. There is a risk that the differing persistence in activity between the products could affect the FEC seen. Ivermectin has a longer duration of activity and thus may be effective against some level of reinfection from parasites overwintering on pastures. Fenbendazole does not have any residual activity and it may appear that fenbendazole is not as effective simply due to this difference.
Mackie KG, Menzies PI, Bateman KG, Gordon JL. Efficacy of fenbendazole and ivermectin in treating gastrointestinal nematode infections in an Ontario cow-calf herd. Can Vet J. 2019 Nov;60(11):1213-1219. PMID: 31692638; PMCID: PMC6805021.
Efficacy of Fenbendazole and Ivermectin against Trichuris spp. in African Green Monkeys (Chlorocebus sabaeus) in Barbados West Indies (2021)
In conclusion, both fenbendazole and ivermectin are effective anthelmintics in treating Trichuris spp. infection in African green monkeys. All treatment groups showed significant reductions in FEC when compared with baseline counts and control animals; however, fenbendazole may be more effective than ivermectin when used solely or in combination with other anthelmintic treatments.
…Fenbendazole has been administered at doses of 10 to 50 mg/kg PO daily for 3 to 5 d, with or without repeated doses.16,38,44,45 Ivermectin has been administered at doses of 100 to 400 µg SC, IM, or PO as a single dose or at specified intervals.21,24,38,51 Little published data exist regarding the use of these drugs specifically in AGM. The current study was conducted to compare the efficacy of fenbendazole and ivermectin—alone and in combination—against Trichuris spp. in naturally infected AGM in Barbados, West Indies, for clearance of this parasite.
…in this study, both fenbendazole alone and combined with ivermectin proved to be more effective than use of ivermectin alone for the treatment of Trichuris spp. in AGM. Concurrent treatment with fenbendazole and ivermectin appeared to be the most effective treatment, based on the rapid decrease in shedding rates and FEC. Studies in other Old World primate species similarly show that avermectins are less effective than benzimidazoles in treating Trichuris spp. infections.
Figure 2. Estimated mean fecal egg counts (FEC) for the fenbendazole (FBZ), ivermectin (IVM), and combined (FBZ+IVM) treatment groups and the control group over time. Note that day 0 values are pretreatment mean FEC. Error bars represent 95% CI.
Rhynd K, Walsh DP, Arthur-Banfield LC. Efficacy of Fenbendazole and Ivermectin against Trichuris spp. in African Green Monkeys (Chlorocebus sabaeus) in Barbados West Indies. J Am Assoc Lab Anim Sci. 2021 Jul 1;60(4):475-483. doi: 10.30802/AALAS-JAALAS-20-000103. Epub 2021 May 10. PMID: 33972010; PMCID: PMC9390614.
A discussion piece for licensed medical practitioners only. Not an endorsement or promotion. Not checked for efficacy, safety or interactions:
Ivermectin vs. Fenbendazole for Cancer: Differences and Which Is Better? (2022)
By Cameron-Leigh Henning
In recent years, unconventional drugs like Fenbendazole and Ivermectin have gained popularity as alternative forms of treatment for cancer. We’ve broken down the essential things you need to know about these medications for cancer in humans.
What Is Ivermectin?
Ivermectin is an anti-parasite drug used to treat various parasite infections in humans, but it can also be used to treat other health conditions like river blindness, onchocerciasis, intestinal strongyloidiasis, and onchocerciasis.
What Is Fenbendazole?
Fenbendazole is a benzimidazole that offers a wide spectrum anthelmintic effect. It is a dewormer medication used to treat parasites in dogs and cats. Some common names for Fenbendazole are Panacur® and Safe-Guard®.
The Role of Ivermectin in Cancer Symptoms
Several clinical trials have been done to prove the effectiveness of Ivermectin against cancer cells with low enough dosages to be non-toxic to the normal cells. Research suggests that Ivermectin suppresses the growth and spread of cancer cells and promotes cancer cell death. Ivermectin proved successful against cancer cells when combined with chemotherapy or other targeted drugs and shows brilliant effectiveness against conventional chemotherapy drug-resistant cancer cells.
Ivermectin also shows efficacy for colorectal antitumor properties. Colorectal cancer still doesn’t have an effective treatment, but Ivermectin has been shown to possess anti-virus, anti-inflammatory, and antitumor properties. Another study shows that after treatment with Ivermectin for breast cancer, the proliferation of multiple breast cancer cell lines was significantly reduced.
The Role of Fenbendazole in Cancer Symptoms
Fenbendazole acts as a moderate microtubule destabilizing agent and possesses a potent antitumor effect. Fenbendazole causes cancer cells to erupt by modulating multiple cellular pathways. To prove the effectiveness of using fenbendazole for cancer, a study treated human non-small cell lung cancer with this dewormer. The cancer cells were analyzed, and researchers found that fenbendazole causes partial alteration of the microtubule network around the nucleus of the cell. There was also an increase in the WT p53 tumor suppressor genes, which enhanced cell death-inducing activity.
Cancer cells have been shown to use more glucose to fuel their energy requirements. Researchers tested the effectiveness of fenbendazole on glucose uptake in human cancer cells and found inhibition of glucose in the cell lines. Researchers also suggest that fenbendazole can be beneficial in evading the drug resistance encountered in cancer therapy.
Should You Use Ivermectin or Fenbendazole for Cancer?
Ivermectin Effectiveness
Ivermectin and breast cancer Ivermectin has been found to turn cold breast tumors hot. Cold tumors mean there are little to no infiltrating T-cells. However, Ivermectin treatment led to robust T-cell infiltration, which turned the tumors into hot ones. This suggests that Ivermectin could synergize with proteins like the PD-1, which help the immune system by acting as a brake on T-cells. This will increase immunity and help the body eradicate cancer.
Ivermectin and digestive system cancer Clinical studies show that dose-dependant Ivermectin inhibits the proliferation of glioblastoma cells in humans and induced apoptosis. Ivermectin has the potential to resist tumor angiogenesis and tumor metastasis.
Ivermectin and lung cancer Ivermectin significantly inhibits the production of lung cancer cells by inhibiting the YAP1 activity. Ivermectin can also reduce the metastasis of lung cancer cells by impeding EMT.
Ivermectin and melanoma Melanoma cells were treated with Ivermectin and showed the potential to effectively inhibit melanoma activity.
Ivermectin and ovarian cancer Ivermectin has the potential to block a cell cycle and induce cell apoptosis in ovarian cancer. The combination of Ivermectin and paclitaxel has a synergized effect on ovarian cancer. A combined treatment of these two almost completely inhibited tumor growth in vivo.
Ivermectin and colon cancer Ivermectin has been shown to have anti-virus, anti-inflammatory, and antitumor properties when it comes to colorectal cancer.
Fenbendazole Effectiveness
Fenbendazole and prostate cancer Fenbendazole has been shown to be cytotoxic against paclitaxel-resistant prostate cancer.
Fenbendazole and lung cancer Fenbendazole has been found to be effective against small cell lung cancer, and one of the success stories of this is Joe Tippens, who had small cell lung cancer that spread to his neck, right lung, stomach, liver, bladder, pancreas, and tail bone—and he was given three months to live. He managed to cure his cancer with a mix of fenbendazole, curcumin, CBD oil, and vitamin E, and has been cancer-free ever since.
Fenbendazole and lymphoma A recent study shows that a combination of fenbendazole and supplemented vitamins inhibited the growth of human lymphoma cells in mice.
Overall, when it comes to cancer in the body, fenbendazole has been shown to destroy the tubular structure of the cancer cells, reduce the ability of cancer cells to metabolize, and boost the body’s immune response to the cancer cells.
In Summary: Which Product Is Better?
It is impossible to say which product is more effective as both are alternate forms of treatment for cancer, and their effectiveness is still being studied—despite the promising results and relevant success stories.
And for an objective balanced review, this report on the “fenbendazole scandle”:
How cancer patients get fake cancer information: From TV to YouTube, a qualitative study focusing on fenbendazole scandle (2022)
Background: Korean society has faced challenges in communicating with cancer patients about false information related to complementary alternative medicine. As the situation has become severe with the 2020 fenbendazole scandal, the demand for reliable information from health authorities has increased.
Objectives: This study aimed to examine patients’ acquisition patterns and perception of false information by presenting empirical evidence to help health authorities enable effective preemptive responses in the cancer communication context.
Method: We conducted a focus group interview with 21 lung cancer patients who were informed about fenbendazole based on a semi-structured questionnaire with three categories: 1) acquisition channel of the general cancer information and the false information, 2) quality of obtained information, and 3) perception toward it. The interviewees, comprising 13 men and eight women, were aged 50 or older. Participants’ current stages of cancer were stages one, three, and four and there were seven people in each stage.
Results: 1) Acquisition channel: Participants had their first encounter with false information through the TV, while the channels to obtain general cancer information were through Internet communities or portal sites. YouTube was a second channel to actively search for information regardless of the information type. 2) Information quality: participants had only fragmented information through media. 3) Perception: Most patients had a negative attitude toward complementary and alternative medicine information such as fenbendazole. They perceive that it needs to be verified by experts and filtered according to their arbitrary criteria. They had vague expectations based on a hope for “what if” at the same time.
Conclusions: Despite the complex media environment, traditional or legacy media is an important channel to encounter information. YouTube is independent of other media as an “active” information-seeking channel. Patients required the appropriate intervention of experts and governments because they perceived that they had obtained irrational and unreliable information from the media. Suggestions are made about how health authorities can construct an effective communication system focusing on the user to prevent patients from getting false cancer information.
Despite the growing number of channels to obtain information, patients feel that there is a lack of cancer information as CAM information generally does not provide scientific evidence and there is little information that they need (A, B, D, G, K, L, N, O, and P). Participants explained that the information on how to take care of themselves, especially after surgery or chemotherapy, when staying at home for self-care and boosting immunity is not sufficient (A-U). T said, “The hospital provides treatment, but after that, most of us are at home. Doctors do not talk about how to manage at home and I cannot get reliable information through the media.”
All participants hoped that experts would come forward with cancer information (A-U). They mentioned it would be good for medical staff or officials to sort out this confusing situation because only experts can verify and deliver reliable information. B explained, “We know that cancer gets better or worse depending on how doctors treat it. But, how should I take care of my body so that it does not happen again? We need this information. I wish doctors or other hospital staff could teach me.” Most people mentioned the experts’ intervention for CAM information related to dietary therapy. J said that it would be better if the doctor gave an example of the foods that are good for my body and which should be avoided rather than saying, “Eat well.” P said, “If you look on the Internet, some say you should eat red ginseng, and some say you should not. What should I do to boost my immunity? I need such information, and I hope the experts will tell me.”
More:
Kim JH, Oh KH, Shin HY, Jun JK. How cancer patients get fake cancer information: From TV to YouTube, a qualitative study focusing on fenbendazole scandle. Front Oncol. 2022 Oct 28;12:942045. doi: 10.3389/fonc.2022.942045. PMID: 36387110; PMCID: PMC9650234.
From 2022, Wang et al published a literature review investigating the effects of quercetin on lncRNAs :
The Targeting of Noncoding RNAs by Quercetin in Cancer Prevention and Therapy (2022)
It has well documented that quercetin exerts anticancer actions by inhibiting cell proliferation, inducing apoptosis, and retarding the invasion and metastasis of cancer cells. However, the exact mechanism of quercetin-mediated cancer chemoprevention is still not fully understood. With the advances in high-throughput sequencing technologies, the intricate oncogenic signaling networks have been gradually characterized. Increasing evidence on the close association between noncoding RNA (ncRNAs) and cancer etiopathogenesis emphasizes the potential of ncRNAs as promising molecular targets for cancer treatment. Available experimental studies indicate that quercetin can dominate multiple cancer-associated ncRNAs, hence repressing carcinogenesis and cancer development. Thus, modulation of ncRNAs serves as a key mechanism responsible for the anticancer effects of quercetin.
Biological functions:
Existing evidence has proven that the antioxidant activity of quercetin is predominantly manifested through its impact on glutathione (GSH), antioxidant enzymatic activity, reactive oxygen species (ROS), and various signaling cascades (e.g., NF-κB, AMPK, and PI3K/Akt) [32]. GSH can defend against oxidative stress by acting as a hydrogen donor in the neutralization of hydrogen peroxide [33]. In vivo experimental results showed that high intake of quercetin significantly enhanced GSH production in the liver [34]. Quercetin also stimulated the expression of the antioxidant enzymes glutathione peroxidase 1 (GPX1), catalase (CAT), and superoxide dismutase 1 (SOD1) in the liver by activating the nuclear factor E2-related factor 2 (Nrf2) pathway. Oppositely, quercetin exhibited a powerful suppressive effect against key enzymes with oxidative activities, such as acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) [35]. Thus, quercetin functions to strengthen the antioxidant defense system.
Quercetin has strong antioxidant and free radical scavenging capacities by removing ROS and inhibiting lipid peroxidation [36]. It is known that iron is a life-supporting micronutrient that is dispensable in the human diet and plays a crucial role in various fundamental metabolic processes. However, excessive iron accumulation can cause cell and tissue damage, since highly reactive iron catalyzes ROS generation. Quercetin effectively reduces iron deposition to inhibit lipid peroxidation and eliminates iron-catalyzed ROS production [37]. Based on the above evidence, quercetin exerts iron-chelating and ROS-scavenging activities.
…It has been well documented that quercetin exhibitspowerful anti-inflammatory effects and attenuates the inflammation process. The immunosuppressive capacities of quercetin are mediated by its effects on the levels of inflammatory cytokines, the generation of inflammation-producing enzymes (cyclooxygenase (COX) and lipoxygenase (LOX)), the maintenance of the stability of mast cells, and functional properties of immune cells (e.g., peripheral blood mononuclear cells and T cells) [2]. Quercetin showsbroad-spectrum antimicrobial properties. It was reported that quercetin had a significant inhibitory effect on the growth of pathogenic microbes such as Aspergillus flavus, Candida albicans, Escherichia coli, Listeria, Proteus, Pseudomonas aeruginosa, Salmonella enteritidis, Shigella, and Staphylococcus aureus [41–43]. The antimicrobial mechanisms of quercetin involve the destruction of bacterial cell wall/membrane, alternation of cell permeability, regulation of protein synthesis and abundance, inhibition of enzymatic activities, and nucleic acid biosynthesis.
Circular RNA (or circRNA) is a type of single-stranded RNA which, unlike linear RNA, forms a covalently closed continuous loop. In circular RNA, the 3' and 5' ends normally present in an RNA molecule have been joined together. This feature confers numerous properties to circular RNA, many of which have only recently been identified.
Many types of circular RNA arise from otherwise protein-coding genes. Some circular RNA has been shown to code for proteins.[1][2] Some types of circular RNA have also recently shown potential as gene regulators. The biological function of most circular RNA is unclear.
Because circular RNA does not have 5' or 3' ends, it is resistant to exonuclease-mediated degradation and is presumably more stable than most linear RNA in cells.[3] Circular RNA has been linked to some diseases such as cancer.[4]
This is a representative figure showing mature mRNA splicing and corresponding canonical biogenesis of circRNAs which might include B.1) direct back splicing. B.2) Intron pairing driven circularization, B.3) debranching resistant intron lariat and B.4) lariat-driven circularization (exon skipping). https://en.wikipedia.org/wiki/Circular_RNA#/media/File:Circular_RNA_Biogenesis.jpg
Quercetin and its derivatives modulate a wide range of cancer related noncoding and long noncoding RNAs:
CCAT1 or Colon Cancer Associated Transcript 1 is a lncRNA that promotes tumor formation and is upregulated in colon cancer and other cancer cell types.
MALAT1 or Metastasis Associated Lung Adenocarcinoma Transcript 1 is perhaps the best known and most significant lncRNA that it interacts with. "It may act as a transcriptional regulator for numerous genes, including some genes involved in cancer metastasis and cell migration, and it is involved in cell cycle regulation. Its upregulation in multiple cancerous tissues has been associated with the proliferation and metastasis of tumor cells".
LncRNAs ENST00000313807 and ENST00000449307 are co-expressed with miRNA LRG1, which play important roles in anti-colorectal cancer mechanisms of quercetin, through competitively binding with miR-5096.
Protein coding MYO10 or Myosin X functions as an actin-based molecular motor and plays a role in the integration of F-actin and microtubule cytoskeletons during cellular meiosis (cell division).
And protein coding ARPP19 or CAMP Regulated Phosphoprotein 19 is a phosphatase inhibitorthatplays a role in regulating mitosis (where replicated chromosomes are separated into two new nuclei)
. “…ablation of this protein in mouse embryonic fibroblasts promotes the premature dephosphorylation of mitotic substrates resulting in the disruption of the correct temporal order of cellular events during mitotic progression”.
Targeting multiple cancer pathways makes it less likely that mutated cells will be able to escape its inhibitory effects, unlike certain allopathic chemotherapeutics that are designed to target just one or so pathways and are often associated with undesirable off-target side effects, thus need to be used in combination:
Henceforth, multitargeted therapy or combination therapy is more superior to single-agent therapy in most cancer treatments. Additionally, combination chemotherapy agents with different mechanisms of action and also nonoverlapping toxicities can be chosen to decrease the resistance and toxicities
Anticancer effects of quercetin and its derivatives by regulation of ncRNAs. ncRNAs participate in key processes during cancer development and progression by modulating their target genes or affecting the activity of cancer-related signaling cascades. Thus, quercetin plays an important role in controlling carcinogenesis, cancer cell proliferation, apoptosis, chemosensitivity, migration, and invasion via alteration of the expression and function of cancer-relevant ncRNAs. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9155922/figure/fig2/
Next we shall go through these in detail, walking through this key section of the review:
7.6. Anticancer Actions of Quercetin Mediated by lncRNAs and circRNAs
Phytochemicals, including curcumin, quercetin, and resveratrol, have emerged as important regulators of lncRNAs in different types of cancers[6]. Hyperoside is a flavonol glycoside compound and exhibits anticancer activities against various cancers [118].
Hyperoside is the 3-O-galactoside of quercetin. A glycoside it is mainly present in plants of the Hypericum and Crataegusgenera, as well as a variety of fruits and vegetables, including onions.
Protein coding FOXO1 or Forkhead Box O1 is thought to play a part in myogenic growth and differentiation. It is the main target of insulin signaling and regulates metabolic homeostasis in response to oxidative stress or starvation, where it can induce autophagic cell death and apoptosis. In the light of this and their effects on angiogenesis and cancer metabolism FOXOs are generally considered to be tumour suppressors:
Foxo1 played a key role in cancer cell apoptosis. The expression level of Foxo1 was decreased, while that of lncRNA colon cancer associated transcript 1 (CCAT1) was elevated in non-small-cell lung cancer (NSCLC) cells. Hyperoside was able to enhance Foxo1 expression by downregulating CCAT1 in NSCLC cells. Further investigation showed that hyperoside repressed the proliferation and promoted the apoptosis of NSCLC cells via the CCAT1-mediated Foxo1 signaling.
Significant downregulation of oncogenic MALAT1. I checked out the referenced in vitro study [21] of treated prostate cancer cells and they used a concentration of 10mM in DMSO, which is very high in comparison to measured peak human plasma concentrations of .43uM (431 nmol/L).
Mean (bold line) and individual (fine line) plasma concentration-time curves of quercetin after oral intake of 50 (A), 100 (B), or 150 mg (C) quercetin to a subgroup of 15 volunteers (n = 5 per dosage group). The last 2 data points are connected by a dashed line, because elimination processes during this time cannot be reliably determined. https://www.sciencedirect.com/science/article/pii/S0022316622099205?via%3Dihub
However, its more complicated than that as long term area under the curve with repeat dosing, IV administration, co-administration, concentration/accumulation in tumour tissues or presence of metabolites of quercetin also need consideration. Low plasma may mean its been absorbed and your MALAT1 is being knocked down regardless.
INTRAVENOUS QUERCETIN: Intravenous Quercetin has studied potential for increased bioavailability [1,2,4] as well as potent potential anti-tumor activity [4-5]. Intravenous data in human subjects shows it to be tolerated and safe [1-3]. Data available suggest multiple mechanisms of action in Tyrosine Kinase inhibition [4] as well as tumor growth suppression [5]. Two years of clinical use has revealed no adverse events when used under standard dose and administration guidelines [3].
This is why in vivo and clinical trials are needed urgently:
The expression level of MALAT1 was remarkably diminished in quercetin-treated prostate cancer cells [21]. MALAT1 played an oncogenic role in cancer progression. Mechanistically, MALAT1 promoted the EMT process in prostate cancer cells by upregulating N-cadherin and downregulating E-cadherin. Moreover, MALAT1 activated the PI3K/Akt signaling cascade by increasing the expression level of phosphorylated Akt. Quercetin inhibited cell proliferation, invasion, and migration but facilitated apoptosis in prostate cancer cells via blocking the EMT process and the PI3K/Akt signaling cascade by targeting MALAT1. Oppositely, MALAT1 overexpression conferred chemoresistance to prostate cancer cells against the cytotoxicity of quercetin. Thus, MALAT1 could antagonize the tumor-suppressive effects of quercetin. These results suggested that MALAT1 could be a pivotal target in quercetin treatment of prostate cancer, providing a novel molecular basis for the clinical use of quercetin in treating prostate cancer.
Protein coding gene LRG1 for Leucine Rich Alpha-2-Glycoprotein 1 is involved with protein to protein interaction, signal transduction (eg JAK-STAT signalling pathways or biochemical cascades), cell adhesion, granulocyte differentiation, and neovascularisation of damaged cells or in cancer tissues via transforming growth factor beta (TGFb) signalling to endothelial cells:
Quercetin inhibited the proliferation and induced the apoptosis of colon cancer cells by regulating the expression of several lncRNAs (Figure 2) [113]. Quercetin had the ability to downregulate leucine-rich α-2-glycoprotein-1 (LRG1), which might explain its role as an anticancer compound.The lncRNAs ENST00000313807 and ENST00000449307 were able to increase LRG1 expression through competitive interaction with miR-5096. It was likely that quercetin induced colon cancer cell apoptosis by inhibiting LRG1 expression via downregulating ENST00000313807 and ENST00000449307. In addition, the circRNA (8: 93786223|93822563) was predicted to regulate LRG1 expression by competitively combining with miR-5096 [113]. The role and mechanisms of action of the circRNA/miRNA/LRG1 axis in quercetin-mediated tumor inhibition deserve further investigation.
Quercetin can suppress the proliferation of cervical cancer cells via interactions with a wide range of oncogenes, an androgen receptor, miRNAs, ncRNAs and lncRNAs including MALAT1 and 71 circRNAs:
Quercetin suppressed the proliferation of cervical cancer cells [119]. The combined analyses of the Gene Expression Omnibus (GEO) database and RNA-sequencing results indicated that quercetin altered the expression of several genes, including Jun protooncogene (JUN), androgen receptor (AR), and EGFR. Further exploration of the upstream interacting ncRNAs of these genes identified one lncRNA (MALAT1) and seventy-one circRNAs (e.g., hsa_circ_001859, hsa_circ_0089761, MYO10, and ARPP19). The lncRNA/circRNA-miRNA-mRNA regulatory networks might take part in the antagonistic effects of quercetin against cervical cancer. These results might help provide valuable diagnostic biomarkers and therapeutic targets for cervical cancer. However, it is of great necessity to determine the genuine roles and clinical significance of the identified ncRNAs in cervical cancer.
Finally, albeit the potential of quercetin to treat cancer, its poor bioavailability and the requirement for high doses have a negative impact on its clinical efficacy. As nano-based formulations can enhance the bioavailability and particular targeting of natural compounds, their use may attenuate the dosage and improve the therapeutic effectiveness of quercetin [120]. Collectively, bioactive quercetin is widely available and has various health benefits with limited side effects. Quercetin holds great potential to become an effective agent for cancer prevention and treatment or an adjuvant agent in combination with conventional anticancer therapies. Quercetin is capable of targeting various ncRNAs that need to be further deciphered to adequately understand how this bioactive compound could be useful in cancer intervention.
Pathology linked to the galectin-3 (gal-3) “galectin fold” of spike protein is of particular interest to me and I wrote a Substack on this.
Gal-3 is associated with cancer promotion and metastasis. Block the gal-3 and you may slow down proliferation and help other meds to work.
TME: Tumour microenvironment.
CML: Chronic myeloid/myelogenous leukaemia or chronic granulocytic leukaemia.
Function of extracellular galectin-3 in the TME
Notably, several studies have shown that the behavior of tumor-stromal cells, including endothelial cells, immune cells, cancer-associated fibroblasts, myofibroblasts and MSCs, is affected by the extracellular expression of galectin-3, whereas it has been found that these cells may also secrete galectin-3 (5,79–82). Upregulation of the galectin-3 expression increases the ability of cancer cell migration and invasion in several tumors, including breast, melanoma, lung, sarcoma, gastric cancer and CML (12,35,82–85). In addition, galectin-3 interacts with ECM glycoproteins, such as fibronectin, collagen IV, elastin and laminin, which play pivotal roles in cell migration (86–89). Studies have shown that galectin-3 also interacts with epidermal growth factor receptor (EGFR) to induce its phosphorylation and re-localization from the membrane to the cytoplasm. In the case of colon cancer cell migration, extracellular galectin-3 may bind with EGFR to affect EGFR dynamics (90).
Roles of galectin‑3 in the tumor microenvironment and tumor metabolism (Review)” (2020)
Quercetin is one of the more effective Gal-3 inhibitors, a side effect being inhibition of another anti-cancer pathway.
Highlights on the Role of Galectin-3 in Colorectal Cancer and the Preventive/Therapeutic Potential of Food-Derived Inhibitors, (2022)
Simple Summary
Colorectal cancer (CRC) incidence is increasing worldwide and represents an important health problem.Therapy failure and progression to metastatic disease are major concerns. Among the factors involved in tumor growth, galectin-3 (Gal-3) plays an important role due to its ability to finetune a number of molecular players that act at different levels of cancer-related processes. A clear relationship between Gal-3 and CRC has been demonstrated. Several studies have, indeed, reported a pathogenetic role for this protein in intestinal inflammation and CRC onset/progression. Moreover, some plant-source food-derived bioactive compounds (mostly fibers and polyphenols) can contribute to the control of CRC onset/growth through their capacity to block Gal-3 activities. In this review, we summarize these studies, highlighting the influence of Gal-3 on CRC risk/progression, cancer cell spreading and patient prognosis, as well as the potential of natural food-derived Gal-3 inhibitors as promising candidates for CRC prevention and therapy.
Interesting to see resveratrol and berberine too.
Refs 140-143:
“The apoptotic effect of resveratrol in ovarian cancer cells is associated with downregulation of galectin-3 and stimulating miR-424-3p transcription”, (2019)
“Berberine alleviates oxidized low-density lipoprotein-induced macrophage activation by downregulating galectin-3 via the NF-κB and AMPK signaling pathways”, (2018)
Among polyphenols, resveratrol, a compound attracting considerable interest during the last few decades as an antitumor agent, was found to downregulate Gal-3 expression in ovarian cancer cells through the induction of mir-424-3p [140].
Gal-3 blocking resulted in the inhibition of cancer cell migration and invasion, and in their increased sensitivity to chemotherapy [140].
In a different experimental disease model, HFD-induced atherosclerosis, Li H and colleagues demonstrated that the flavonoid quercetin alleviates atherosclerotic lesions by inhibiting HFD-induced Gal-3 expression and consequent NLRP3 inflammasome activation in macrophages [141].
The negative effect of quercetin on the Gal-3-NLRP3 pathway was also confirmed in in vitro stimulated macrophage cell lines [141]. Accordingly, treatment with berberine suppressed oxidized low-density lipoprotein-induced upregulation of Gal-3 and macrophage activation [142].
Quercetin and berberine were both shown to exert beneficial effects on intestinal inflammation and to inhibit colon tumorigenesis [144,145].
Finally, the effects of xanthohumol and 8-prenylnaringenin, belonging to the prenylflavonoid family, on Gal-3 expression were investigated in a mouse model of type 2 diabetes mellitus [143]. HFD-induced diabetes resulted in overexpression of Gal-3 in the liver and kidney that was associated with oxidative stress. Oral intake of both polyphenols completely suppressed Gal-3 expression and oxidative stress markers [143]. Of note, these compounds have been reported to affect mitochondrial function and oxidative pathways in CRC [146].
Although the effects of polyphenols in regulating Gal-3 activity have been poorly investigated in cancer, the results achieved in different disease models point to Gal-3 inhibition as a novel mechanism underlying the anti-inflammatory and anti-tumoral activity of these compounds.
Polyphenol-mediated effects on Gal-3 have not yet been specifically explored in CRC models, but the observation that Gal-3 is highly expressed in CRC tissues, together with its known role in intestinal inflammation and colon carcinogenesis, suggest that the capacity of these compounds to modulate Gal-3 in other disease contexts might also be exploited in the management of CRC, thus, further strengthening the potential of their supplementation for therapeutic and prevention purposes.
Data is mixed from the few trials that have been conducted, and long term prevention has not been assessed. More research and delivery systems need to be developed:
Galasso et al (2013) administered either 500 mg/d of quercetin, 100 mg/d of genistein or placebo to 18 men with elevated prostate-specific antigen (PSA), a marker for prostate cancer, over 3 months.
800 CHEMOPREVENTIVE PROPERTIES OF THE FLAVONOIDS GENISTEIN AND QUERCETIN IN MEN WITH RISING PSA: FIRST RESULTS OF A DOUBLE-BLIND, RANDOMIZED, PLACEBO-CONTROLLED TRIAL (QUERGEN TRIAL). (2013)
INTRODUCTION AND OBJECTIVES
Polyphenols are known to have beneficial effects in the chemoprevention of prostate cancer. Several in vitro and in vivo studies demonstrated the anticancerogenic action of the flavonoids quercetin and genistein. However, human interventions still have to prove that these substances have chemopreventive efficacies. Therefore, this study aims to investigate the effect of genistein and quercetin supplementation on the concentration and dynamics of prostate-specific antigen (PSA) in men with rising PSA-levels.
METHODS
The first interim analysis of the ongoing QUERGEN trial included a total of 18 men with elevated PSA concentrations. The subjects were randomized to receive either 500 mg/d of quercetin (Q), 100 mg/d of genistein (G) or placebo (P) for 3 months. Serum PSA as primary endpoint and bioavailability of quercetin and genistein through HPLC analyses were assessed prior to intervention and after the supplementation period.
RESULTS
The blood quercetin concentration increased significantly in the quercetin intervention group (pre: 0.5±0.07 μmol/l, post: 1.2±0.05 μmol/l, p=0.0313). Genistein supplementation also tended to increase genistein levels (pre: 1.1±0.05 μmol/, post: 1.3 ±0.06 μmol/l, ns). Total and free PSA did not change significantly in none of the groups (Q: pre tPSA 8.9±2.9 ng/ml, post 10.5±3.8 ng/ml, ns; pre fPSA: 0.8±0.2 ng/ml, post: 1.0±0.3 ng/ml; ns; G: pre tPSA: 9.5±2.3 ng/ml, post: 9.6±2.2 ng/ml; ns; pre fPSA 0.7±0.1 ng/ml, post: 0.8±0.3 ng/ml, ns; P: pre tPSA: 6.3±0.8 ng/ml, post: 7.4±0.8 ng/ml, ns; pre fPSA 0.9±0.2 ng/ml, post: 0.8±0.1 ng/ml, ns). However, there was a trend to an inverse association between the PSA dynamics and the intake of genistein. After intervention PSA doubling time was higher in the genistein intervention group compared with quercetin and placebo receiving subjects (G: 16.2±7.8 months, Q: 7.0±2.8 months, P: 7.5±1.8 months, ns). Furthermore, the log2-transformated PSA slope was 0.08 ng/ml in the genistein group and thus lower compared to the other groups (P: 0.25 and Q: 0.20 ng/ml, ns).
CONCLUSIONS
In this interim analysis, the results suggest that dietary intervention with genistein may have an impact on PSA slope in men with deviant PSA constellation suspicious for prostate cancer. In contrast, the intake of quercetin did not impact on PSA dynamics. Within the QUERGEN trial more patients with longer intervention periods are currently recruited to verify these initial results.
In contrast quercetin administered by IV to ovarian cancer patients demonstrated antitumor activity.
Tyrosine kinases are enzymes that are found at high levels in some types of cancer, and inhibition can stop cancer cells from growing. They are a part of many cell functions including cell signalling, growth and division.
From Ferry et al:
Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition, (1996)
Abstract
We have performed a Phase I clinical trial with the naturally occurring flavonoid quercetin (3,3',4',5,7-pentahydroxyflavone). Quercetin has antiproliferative activity in vitro and is known to inhibit signal transduction targets including tyrosine kinases, protein kinase C, and phosphatidyl inositol-3 kinase. Quercetin was administered by short i.v. infusion at escalating doses initially at 3-week intervals. The first dose level was 60 mg/m2; at the 10th dose level of 1700 mg/m2, dose-limiting nephrotoxicity was encountered, but no myelosuppression. At the preceding dose level of 1400 mg/m2, five patients were treated at 3-week intervals, and another eight patients were treated on a once-weekly schedule; overall, 2 of 10 evaluable patients had renal toxicity, 1 at grade 2 and 1 at grade 4. We therefore treated other patients at 945 mg/m2 (eight at 3-week intervals and six at weekly intervals); 3 of 14 patients had clinically significant renal toxicity, 2 patients with grade 2 and 1 patient with grade 3. Patients treated on the weekly schedule did not have cumulative renal impairment but did have a fall in the glomerular filtration rate of 19 +/- 8% in the 24 h after drug administration. We recommend 1400 mg/m2 as the bolus dose, which may be given either in 3-week or weekly intervals, for Phase II trials. Quercetin pharmacokinetics were described by a first-order two-compartment model with a median t(1/2)alpha of 6 min and median t(1/2)beta of 43 min. The median estimated clearance was 0.28 liter/min/m2, and median volume of distribution at steady state was 3.7 liter/m2. In 9 of 11 patients, lymphocyte protein tyrosine phosphorylation was inhibited following administration of quercetin at 1 h, which persisted to 16 h. In one patient with ovarian cancer refractory to cisplatin, following two courses of quercetin (420 mg/m2), the CA 125 had fallen from 295 to 55 units/ml, and in another patient with hepatoma, the serum alpha-fetoprotein fell. In conclusion, quercetin can be safely administered by i.v. bolus at a dose injection. The plasma levels achieved inhibited lymphocyte tyrosine kinase activity, and evidence of antitumor activity was seen.
More on clinical trials and other data from an article by Hein on Medscape:
Any Merit to Quercetin's Anticancer Claims?, (2020)
But do these preclinical results translate to anticancer effects in humans?
Unfortunately, clinical trials addressing this question are lacking. Several observational studies evaluating the link between dietary flavonoid intake and cancer have been published, with many showing an inverse association. The best evidence looking specifically at quercetin intake comes from a 2013 pooled analysis of cohort and case-control studies by Woo and Kim. These researchers found that a higher intake of quercetin was associated with 20% lower odds of cancer, with an even greater risk reduction in smokers. However, individual studies addressing various forms of cancer show mixed results. For instance, an inverse association between quercetin intake and both pancreatic cancer and gastric cancer has been reported, whereas studies on bladder cancer and ovarian cancer risk failed to identify such a link. Observational research on quercetin's effect on lung cancer risk has been particularly conflicting, with separate studies showing no association, a trend toward inverse association, and a 35% lower odds of lung cancer with increased quercetin intake.
We must recognize that such studies cannot address causation, only association, making it difficult to say whether quercetin intake alone was responsible for the reduced cancer risk. Although most of the studies were adjusted to account for potential confounders, including intake of other flavonoids, use of other supplements, physical activity, education, and socioeconomic status, it's impossible to fully account for the known and unknown variables that might influence the results. To gather data for these studies, researchers must also rely on food diaries or the long-term memory of study participants' dietary habits, raising obvious concerns about reliability and recall bias.
Because these studies solely analyzed the dietary intake of quercetin, they tell us nothing about its possible effects when taken in supplement form. And although no safety concerns exist with quercetin from dietary sources, the long-term safety of supplemental quercetin is not well established in humans. Even if quercetin does have anticancer potential, its poor oral bioavailability may make these effects moot, although delivery systems to enhance absorption of quercetin are being investigated.
When discussing the proposed anticancer effects of quercetin, we must also be vigilant in calling out other preclinical research highlighting the possibility for quercetin-related drug interactions. Quercetin has been shown to interfere with several cytochrome P450 (CYP) isoenzymes and drug transporters, including inhibition of CYP2C9, CYP2D6, CYP3A4, organic anion transporting polypeptides, and P-glycoprotein. This suggests that quercetin may alter systemic exposure to a majority of prescription and nonprescription drugs on the market, including anticancer drugs.
Even if the jury is still out on quercetin's anticancer effects, and relevant concerns exist about its bioavailability, safety, and potential drug interactions, the evidence that suggests a lower risk for cancer with dietary intake could potentially be beneficial. While patients may wish to adopt healthier diets with foods known to contain high concentrations of quercetin and other flavonoids, it's too early for healthcare providers to specifically recommend quercetin, particularly in supplement form, for the prevention or treatment of cancer.
If this leads us to conclude anything its to incorporate flavonoids into your diet or via high dose supplements with vitamin C or bromelain for synergistic effects.
And get cooking your German onion cake (as recommended by one of Dr Jo’s followers. It looks delicious! Or even take one of those kale smoothies, yeah🤢.
Quercetin is a strong antioxidant and a major dietary flavonoid. Epidemiological studies suggest that consumption of quercetin protects against cardiovascular disease, but its absorption in man is controversial. We fed nine subjects a single large dose of onions, which contain glucose conjugates of quercetin, apples, which contain both glucose and non-glucose quercetin glycosides, or pure quercetin-3-rutinoside, the major quercetin glycoside in tea. Plasma levels were then measured over 36 h. Bioavailability of quercetin from apples and of pure quercetin rutinoside was both 30% relative to onions. Peak levels were achieved less than 0.7 h after ingestion of onions, 2.5 h after apples and 9 h after the rutinoside. Half-lives of elimination were 28 h for onions and 23 h for apples. We conclude that conjugation with glucose enhances absorption from the small gut. Because of the long half-lives of elimination, repeated consumption of quercetin-containing foods will cause accumulation of quercetin in blood.
Relative bioavailability of the antioxidant flavonoid quercetin from various foods in man, (1997)
Quercetin also improves bioavailability and efficacy of berberine:
% CDR = % cumulative drug release.
…In conclusion, quercetin could be successfully utilized as bioenhancer to improve ex vivo permeability of berberine chloride, which would be expected to improve its bioavailability and reduce the dose resulting in improved patient compliance.
…increased permeability of berberine chloride during pre-treatment study with quercetin might have resulted from the quercetin, which inhibited the efflux pump P-gp. Briefly, inhibition of efflux pump P-gp by quercetin might be duly responsible for permeability enhancement of berberine chloride.
…Based on these data, it could be suggested that 10 mg of quercetin for 30 minutes pretreatment time was optimum to increase the permeability of poorly permeable berberine chloride up to a maximum of 90.91% ± 1.66% CDR.
Quercetin as Natural Bioavailability Modulator: An Overview, (2020)
Its no coincidence that galectin inhibitors tend to make good SARS-CoV-2 inhibitors too:
…The in-silico evaluations were conducted using the PyRx virtual screening tools which lead to the target based on high binding affinity. Trigoneoside IB, derived from Trigonella foenum-graecum (Fenugreek), showed the highest binding affinity and stable interaction with the amino acid residues present in active sites of Covid-19 proteins.
Virtual screening of potential phyto-candidates as therapeutic leads against SARS-CoV-2 infection, (2021)
This one by Sigamani et al. crept under the radar somewhat:
An Oral Galectin Inhibitor in COVID-19—A Phase II Randomized Controlled Trial, (2023)
Background: SARS-CoV-2 vaccines play an important role in reducing disease severity, hospitalization, and death, although they failed to prevent the transmission of SARS-CoV-2 variants. Therefore, an effective inhibitor of galectin-3 (Gal-3) could be used to treat and prevent the transmission of COVID-19. ProLectin-M (PL-M), a Gal-3 antagonist, was shown to interact with Gal-3 and thereby prevent cellular entry of SARS-CoV-2 in previous studies.
Results: PL-M treatment significantly (p = 0.001) increased RT-PCR cycle counts for N and ORF genes on days 3 (Ct values 32.09 ± 2.39 and 30.69 ± 3.38, respectively) and 7 (Ct values 34.91 ± 0.39 and 34.85 ± 0.61, respectively) compared to a placebo treatment. On day 3, 14 subjects in the PL-M group had cycle counts for the N gene above the cut-off value of 29 (target cycle count 29), whereas on day 7, all subjects had cycle counts above the cut-off value. Ct values in placebo subjects were consistently less than 29, and no placebo subjects were RT-PCR-negative until day 7. Most of the symptoms disappeared completely after receiving PL-M treatment for 7 days in more patients compared to the placebo group. Conclusion: PL-M is safe and effective for clinical use in reducing viral loads and promoting rapid viral clearance in COVID-19 patients by inhibiting SARS-CoV-2 entry into cells through the inhibition of Gal-3.
…The efficacy of the study drug (Chewable Tablet Galactomannan, 1400 mg PL-M) was assessed in a randomized, double-blind, placebo-controlled clinical trial in ambulatory patients with mild to moderately severe COVID-19.
Where galactomannan shows up is in Fenugreek seeds. If you like your curries you will be familiar with the sweet, nutty flavour reminiscent of maple syrup and burnt sugar. It adds a sweetness and depth and is another broad spectrum therapeutic.
This investigation was conducted to test the potential of the galactomannan (F-GAL) and aqueous extract (FS-AE) of the Fenugreek seed aqueous to prevent liver and kidney damage extracts in streptozotocin (STZ)-induced T1DM in rats.
…Treatments with both drugs reduced fasting hyperglycemia and improved serum and hepatic lipid profiles in the control and diabetic rats. Additionally, F-GAL and FS-AE attenuated the associated reduction in the mass and structure of the islets of Langerhans in diabetic rats and improved the structure of the kidneys and livers. In association, they also reduced the generation of reactive oxygen species (ROS), lipid peroxides, factor (TNF-α), interleukin-6 (IL-6), and nuclear levels of NF-κB p65, and improved serum levels of ALT, AST, albumin, and creatinine. However, both treatments increased hepatic and renal superoxide dismutase (SOD) in the livers and kidneys of both the control and diabetic-treated rats, which coincided with a significant increase in transcription, translation, and nuclear localization of Nrf2. In conclusion, FS-AE and F-GAL are effective therapeutic options that may afford a possible treatment for T1DM by attenuating pancreatic damage, hyperglycemia, hyperlipidemia, and hepatic and renal damage.
Fenugreek Seed Galactomannan Aqueous and Extract Protects against Diabetic Nephropathy and Liver Damage by Targeting NF-κB and Keap1/Nrf2 Axis, (2022)
…The computational results reveal that the compound galactomannan can be ascribed as potential drug candidate against breast cancer and type 2 diabetes rendered by higher molecular dock scores, stable molecular dynamics (MD) simulations results, and lower binding energy calculations.
Exploring the Therapeutic Ability of Fenugreek against Type 2 Diabetes and Breast Cancer Employing Molecular Docking and Molecular Dynamics Simulations, (2018)
Fenugreek: a naturally occurring edible spice as an anticancer agent, (2009)
Abstract
In recent years, various dietary components that can potentially be used for the prevention and treatment of cancer have been identified. In this study, we demonstrate that extract (FE) from the seeds of the plant Trigonella foenum graecum, commonly called fenugreek, are cytotoxic in vitro to a panel of cancer but not normal cells. Treatment with 10-15 ug/mL of FE for 72h was growth inhibitory to breast, pancreatic and prostate cancer cell lines (PCa). When tested at higher doses (15-20 ug/mL), FE continued to be growth inhibitory to PCa cell lines but not to either primary prostate or htert-immortalized prostate cells. At least part of the growth inhibition is due to induction of cell death, as seen by incorporation of Ethidium Bromide III into cancer cells exposed to FE. Molecular changes induced in PCa cells are: in DU-145 cells: down regulation of mutant p53, and in PC-3 cells up regulation of p21 and inhibition of TGF-β induced phosphorylation of Akt. The surprising finding of our studies is that death of cancer cells occurs despite growth stimulatory pathways being simultaneously up regulated (phosphorylated) by FE. Thus, these studies add another biologically active agent to our armamentarium of naturally occurring agents with therapeutic potential.
MCP is also Gal-3 inhibitor with anti-cancer properties, eg prostate cancer.
Pleiotropic Effects of Modified Citrus Pectin, (2019)
by Eliaz & Raz:
Abstract
Modified citrus pectin (MCP) has a low-molecular-weight degree of esterification to allow absorption from the small intestinal epithelium into the circulation. MCP produces pleiotropic effects, including but not limited to its antagonism of galectin-3, which have shown benefit in preclinical and clinical models. Regarding cancer, MCP modulates several rate-limiting steps of the metastatic cascade. MCP can also affect cancer cell resistance to chemotherapy. Regarding fibrotic diseases, MCP modulates many of the steps involved in the pathogenesis of aortic stenosis. MCP also reduces fibrosis to the kidney, liver, and adipose tissue. Other benefits of MCP include detoxification and improved immune function. This review summarizes the pleiotropic effects of MCP.
…Citrus pectin is a soluble dietary fiber derived from the white pith of citrus fruit peels. Pectin is a large and complex molecule in its natural form, weighing 60–300 kilodalton (kDa), and containing a variable degree of (as much as ~70%) esterification. Pectins are a family of covalently linked galacturonic acid-rich polymers, with three identified central pectic polysaccharides regions: homogalacturonan (HG), rhamnogalacturonan-I (RG-I), and substituted galacturonans (GS). Among the GS is rhamnogalacturonan-II (RG-II), which is distinct from RG-I. RG-II has four types of structurally different oligosaccharides chains composed of 12 kinds of glycosyl residues [1] (Figure 1). Native pectin is not degraded during human digestion, and its large size prevents intestinal absorption [2,3]. However, when citrus pectin is modified (MCP) with a specific pH and heat-controlled enzymatic treatment to yield a product with a low molecular weight of <15 kilodaltons (kDa) and a degree of esterification under 5%, it can be absorbed from the small intestinal epithelium into the circulation [3]. The health benefits of MCP are increasingly recognized and summarized in this review and Table 1.
3. Cancer
Most of the morbidity and mortality associated with cancer is caused by metastasis, which is the migration of cancer from the site of primary tumor growth to distant organs and tissues. The metastatic cascade contains several rate-limiting steps that are modulated by Gal-3 and, in turn, by MCP as well [53]. The first step for neoplastic cells is to survive apoptosis that is associated with the loss of anchorage (anoikis) following escape from the primary tumor and intravasation. Galectin-3 protects cancer cells from anoikis [56,57] by causing a cell cycle arrest at the late G1 phase, which is an anoikis-insensitive point [56]. MCP has been shown to downregulate cyclin B and cdc2 in human prostatic JCA-1 cells [58], which may cause an accumulation of cancer cells in G2/M, thereby inducing apoptosis.
The next rate-limiting step in metastasis involves tumor cell arrest in distant organ microvasculature. Galectin-3 has been shown to mediate metastatic cell adhesion to the endothelium [59,60,61,62,63]. MCP was demonstrated to inhibit tumor cell adhesion to the endothelium as well as cancer cell homotypic aggregation involved in metastatic cell arrest in distant organs and the formation of intravascular metastatic deposits [59,64,65,66,67,68].
The third rate-limiting step in metastasis involves a forking point where tumor cells can either proliferate inside organ microvessels until the metastatic tumor outgrows the blood vessel and invades distant organ parenchyma [69], or extravasate before starting secondary tumor growth. Invasive propensity involves a series of tumor cell interactions with ECM proteins associated with the basement membrane and target organ stroma. MCP has been shown to reduce Gal-3-mediated tumor cell interactions with ECM proteins such as laminin [66]. Also, citrus pectin polysaccharides dose-dependently decreased the invasion through matrigel of human endothelial cells [67], of MDA-MB-231 human metastatic breast carcinoma cells [70], and human buccal metastatic cells [70].
Modified Citrus Pectin as a Potential Sensitizer for Radiotherapy in Prostate Cancer, (2018)
by Conti et al:
Abstract
Background: Radiotherapy is one of the primary therapies for localized prostatic carcinoma. Therefore, there is an emerging need to sensitize prostatic cancer cells to chemotherapy/radiotherapy. Modified citrus pectin (MCP) is an effective inhibitor of galectin-3 (Gal-3), which is correlated with tumor progression, proliferation, angiogenesis, and apoptosis.
Purpose: This study was directed to evaluate the efficacy of combining ionizing radiation (IR) with MCP on PCa cells.
Study design: Effects of treatments on PCa cells survival were evaluated using XTT assay, flow cytometry, and clonogenic survival assay. Expression of selected proteins was estimated using western blotting. Cell motility, migration, and invasion were determined. Contribution of reactive oxygen species production to treatment effects on cell viability was tested.
Results:Radiotherapy combined with MCP reduced viability and enhanced radiosensitivity associated with a decrease in Gal-3, cleavage of the precursor of caspase-3, increased expression of the pro-apoptotic protein Bax, and downregulation of DNA repair pathways, poly-ADP-ribose polymerase, and proliferating cell nuclear antigen. MCP significantly reduced the invasive and migratory potential of PCa cells. Combining sodium pyruvate with MCP and IR mitigated the effect on cell viability.
Conclusion: Our findings demonstrated that MCP sensitized PCa cells to IR by downregulating anti-apoptotic Gal-3, modulating DNA repair pathways, and increasing ROS production. For the first time the correlation between MCP, radiotherapy, and Gal-3 for prostatic cancer treatment was found. In addition, MCP reduced the metastatic properties of PCa cells. These findings provide MCP as a radiosensitizing agent to enhance IR cytotoxicity, overcome radioresistance, and reduce clinical IR dose.
Asprin is a non-steroidal anti-inflammatory drugs (NSAID).
I like to cross check studies on PubMed, regardless of the logic to it. This one surprised me somewhat:
Effects of Aspirin and Indomethacin on Galectin-3, (2005)
By Dabelic & Dumic:
Abstract
Galectin-3, a β-galactoside binding lectin, acts as a strong pro inflammatory signal. Many immunomodulatory drugs affect signaling pathways that comprise transcription factors involved in regulation of galectin-3 gene (LGALS3) expression. The aim of this study was to investigate the effects of non-steroidal anti-inflammatory drugs aspirin and indomethacin on the expression of galectin-3, both on mRNA and protein levels. The human monocytic cell line THP-1 was exposed to various therapeutic concentrations of aspirin and indomethacin for 72 hours. Relative RT-PCR method and the GeneScan analysis software were used for assessing the galectin-3 mRNA level and chemiluminescent-western blot analysis was used for measuring the galectin-3 level. The results showed that galectin-3 is a new target molecule of NSAIDs, which cause reduction of both gene and protein expression of galectin-3, but the intensity and time-course of the changes strongly depend on the kind and concentration of the drugs.
RESULTS
To investigate the effects of NSAIDs on the expression of galectin-3 on both the mRNA and protein levels, we exposed the monocytic cell line THP-1 to the various therapeutic concentrations of aspirin (0.1, 0.5, 1and 2 mmol dm–3) and indomethacin (0.1, 1, 5 and 10mmol dm–3).
The relative amount of mRNA for galectin-3 (i.e.,relative expression ofLGALS3) was determined by theGeneScan Analysis of RT-PCR amplicons for galectin-3 and b-actin.
The lowest applied concentration of aspirin (0.1mmol dm–3) did not affect the expression level of LGALS3 during the first three hours of cultivation. A decrease was observed after 5 hours of cultivation, though it became statistically significant 24 hours after the beginning of the treatment. The level of mRNA continued to fall and it reached (31.1±1.4) % of the initial value after 72 hours of cultivation (Figure 2). When THP-1 cells were exposed to medium (0.5 and 1 mmol dm–3) and high (2 mmol dm–3) therapeutic concentrations of ASA, the level of mRNA for galectin-3 dropped sharply already after the first hour of cultivation (to (59.6±7.6)%, (52.2±1.7)% and (45±3.4)% of the initial value, respectively, Figure2). Prolonged cultivation caused a further decrease of the galectin-3 mRNA level in a dose dependent manner. At the end of the treatment (after 72 hours) the expression of LGALS3 dropped to (25.6±4.8) %, (15.5±0.5)% and (12.3±0.04) % of the expression observed in the control cells, respectively (Figure 2).
From: “Figure 2. Aspirin inhibits expression ofLGALS3in THP-1 cells. THP-1 cells were cultivated for1, 3, 5, 24, 48 and 72 hours with 0.1, 0.5,1 and 2 mmol dm–3aspirin. Following the to-tal RNA isolation and reverse transcription,21 cycles of PCR were performed using fluo-rescent primers for galectin-3 and 14 cyclesof PCR using fluorescent primers forb-actin.RT-PCR amplicons were pooled and separatedby capillary electrophoresis on an AbiPrism310 Genetic Analyzer. Results of the two in-dependent experiments performed in duplicatewere calculated using the GeneScan 2.1 Ana-lysis software and expressed as galectin-3/b-actin RT-PCR amplicon peak areas comparedto the control (x±SD). Statistically significantdifferences (p< 0.05 compared to the con-trols) are marked with asterisks.”, source: https://core.ac.uk/reader/14374990
Galectin-3, ab-galactoside binding lectin, has recently emerged as a new powerful cellular modulator at inflammatory foci. Yet, until now there were no data about the effects of NSAIDs on the expression of either galectin-3 or any other lectin. In the presented study, we have demonstrated for the first time that non-steroidal anti-inflammatory drugs aspirin and indomethacin, applied in clinically relevant doses, effectively inhibited galectin-3 expression at the mRNA level, and to a lesser extent at its protein level in the monocytic cell line THP-1.
Aspirin is also a cyclooxygenase-2 (COX2) inhibitor.
Cyclooxygenase (COX), also known as prostaglandin endoperoxide synthase, is the key enzyme required for the conversion of arachidonic acid to prostaglandins. Two COX isoforms have been identified, COX-1 and COX-2. In many situations, the COX-1 enzyme is produced constitutively (e.g., in gastric mucosa), whereas COX-2 is highly inducible (e.g., at sites of inflammation and cancer). Traditional nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit both enzymes, and a new class of COX-2 selective inhibitors (COXIBs) preferentially inhibit the COX-2 enzyme.
Since its first synthesis in 1897, several medicinal roles and mechanisms of action of aspirin have become apparent; the latest among these being its role in cancer prevention and treatment. A large body of evidence supports aspirin's effect in reducing cancer incidence and cancer mortality, but duration of use needs to be at least 5 years. The beneficial effects are particularly large for colorectal, oesophageal and gastric cancers, with apparently smaller reductions for breast, prostate and lung cancer. The major harm is gastrointestinal bleeding, but serious sequelae are minimal at ages <70 years. It is very likely that use of prophylactic aspirin in the general population aged 50-70+ years will result in net overall benefit. Outstanding issues are: whether standard dose (~300 mg/day) can lead to greater net benefits than low dose (75-100 mg/day), the optimum duration of use, and appropriate ages for use in average-risk individuals.
Low dose is fine, significant but selective anti-cancer effects were found in this large scale cohort analysis from Hong Kong by Tsoi et al:
Long-term use of low-dose aspirin for cancer prevention: A 10-year population cohort study in Hong Kong, (2019)
Abstract
Aspirin, commonly used for prevention of cardiovascular and cerebrovascular diseases, has been found to possess protective effects against cancer development in the Western populations. Such effects among Asian populations remain uncertain. The objective of this study is to investigate the use of aspirin on prevention of different cancers among Chinese users. This population-based study utilized database from the Hong Kong Hospital Authority; adults with aspirin prescription for at least 6 months between 2000 and 2004 were included and followed up until 2013. Aspirin users were age-sex matched with non-aspirin users at a 1:2 ratio. Incidences of cancer were the primary outcome measured by relative risk (RR). A total of 204,170 aspirin users and 408,339 non-aspirin users were included, with the mean age 67.5 years, 7.7 years average duration of aspirin prescription and 80 mg as the median dose of aspirin. Cancer incidences were found in 26,929 (13.2%) aspirin users and 70,755 (17.3%) non-aspirin users. Compared with patients who had not been prescribed aspirin, aspirin usage led to significant reduction of cancers in liver (RR: 0.49), stomach (RR: 0.42), colorectum (RR: 0.71), lung (RR: 0.65), pancreas (RR: 0.54), oesophagus (RR: 0.59) and leukaemia (RR: 0.67). There was no demonstrable reduction of kidney cancer, bladder cancer, prostate cancer and multiple myeloma in association with the usage of aspirin. Risk of breast cancer was shown to marginally increase (RR: 1.14) with aspirin usage.This study demonstrated that the long-term use of low-dose aspirin is associated with the reduction in risk of various cancers but not for breast cancer. Further investigation is needed before promoting aspirin as a primary chemoprotective agent.
Keywords: Chinese; Hong Kong; aspirin; cancer incidence; long-term.
Its particularly useful if you are in certain familial at-risk groups:
Efficacy of Low-Dose Aspirin in Colorectal Cancer Risk Prevention is Dependent on ADH1B and ALDH2 Genotype in Japanese Familial Adenomatous Polyposis Patients, (2022)
by Mure et al:
Abstract
Aspirin has gained great attention as a cancer preventive agent. Our previous study revealed that the low-dose aspirin prevents colorectal tumor recurrence in Japanese patients with colorectal adenomas and/or adenocarcinomas, whereas aspirin increases risks in smokers and has no effects on regular drinkers. Our recent study revealed that aspirin reduces polyp growth in Japanese patients with familial adenomatous polyposis (FAP). In this study, we have studied the association of genotypes of alcohol metabolizing enzymes (ADH1B and ALDH2) on aspirin's efficacy of suppressing polyp growth (≥5 mm) in a total of 81 Japanese patients with FAP. Our study revealed that aspirin showed significant preventive effects for patients with ADH1B-AA and AA+GA types [OR = 0.21; 95% confidence interval (CI), 0.05-0.95, and OR = 0.31; 95% CI, 0.10-0.95, respectively], and for patients with ALDH2-GG and GG+GA types (OR = 0.10; 95% CI, 0.01-0.92, and OR = 0.29; 95% CI, 0.09-0.94, respectively), but not for patients with ADH1B-GG and GA+GG types, and ALDH2-AA and GA+AA types. In addition, substantial preventive effects of aspirin were seen for patients with ADH1B-AA type who do not drink regularly (<3 times/week, OR = 0.11; 95% CI, 0.02-0.78), where a statistically significant interaction between aspirin and ADH1B was observed (Pinteraction = 0.036). Results from this exploratory study strongly indicate that aspirin is beneficial in prevention of polyp growth for patients with FAP with ADH1B-AA and AA+GA types, and ALDH2-GG and GG+GA types. Taken together, we propose ADH1B and ALDH2 as candidate markers for the personalized prevention by aspirin.
Significance: Aspirin is beneficial to patients with FAP with ADH1B-AA and AA+GA types or ALDH2-GG and GG+GA types. ADH1B and ALDH2 genotypes can be the markers for the personalized prevention of colorectal cancer by aspirin.
For editorial balance, an RCT sponsored by Bayer found little correlation between low dose aspirin and cancer incidence. No surprise there. If it did they would have quickly buried it, pardon the expression:
Low-Dose Aspirin in the Primary Prevention of Cancer, The Women’s Health Study: A Randomized Controlled Trial”, (2005)
by Cook et al:
Abstract
Context Basic research and observational evidence as well as results from trials of colon polyp recurrence suggest a role for aspirin in the chemoprevention of cancer.
Objective To examine the effect of aspirin on the risk of cancer among healthy women.
Design, Setting, and Participants In the Women’s Health Study, a randomized 2 × 2 factorial trial of aspirin and vitamin E conducted between September 1992 and March 2004, 39 876 US women aged at least 45 years and initially without previous history of cancer, cardiovascular disease, or other major chronic illness were randomly assigned to receive either aspirin or aspirin placebo and followed up for an average of 10.1 years.
Intervention A dose of 100 mg of aspirin (n=19 934) or aspirin placebo (n=19 942) administered every other day.
Main Outcome Measures Confirmed newly diagnosed invasive cancer at any site, except for nonmelanoma skin cancer. Incidence of breast, colorectal, and lung cancer were secondary end points.
Results No effect of aspirin was observed on total cancer (n = 2865; relative risk [RR], 1.01; 95% confidence interval [CI], 0.94-1.08; P = .87), breast cancer (n = 1230; RR, 0.98; 95% CI, 0.87-1.09; P = .68), colorectal cancer (n = 269; RR, 0.97; 95% CI, 0.77-1.24; P = .83), or cancer of any other site, with the exception of lung cancer for which there was a trend toward reduction in risk (n = 205; RR, 0.78; 95% CI, 0.59-1.03; P = .08). There was also no reduction in cancer mortality either overall (n = 583; RR, 0.95; 95% CI, 0.81-1.11; P = .51) or by site, except for lung cancer mortality (n = 140; RR, 0.70; 95% CI, 0.50-0.99; P = .04). No evidence of differential effects of aspirin by follow-up time or interaction with vitamin E was found.
Conclusions Results from this large-scale, long-term trial suggest that alternate day use of low-dose aspirin (100 mg) for an average 10 years of treatment does not lower risk of total, breast, colorectal, or other site-specific cancers. A protective effect on lung cancer or a benefit of higher doses of aspirin cannot be ruled out.
Financial Disclosures:Dr Cook has served as a consultant to Bayer. Dr Gaziano has served as a consultant to and received grant support from Bayer and McNeil.Dr Ridker has received grant support from Bayer. Dr Hennekens has served as a consultant to Bayer and McNeil and received grant support from Bayer.
Funding/Support: This study was supported by grants HL-43851 and CA-47988 from the National Heart, Lung, and Blood Institute and the National Cancer Institute, Bethesda, Md. Aspirin and aspirin placebo were provided by Bayer Healthcare. Vitamin E and vitamin E placebo were provided by the Natural Source Vitamin E Association.
Awkward, given that they funded all the participants in the past and provided the drugs for the trial. Very generous of them:
Role of the Sponsors: Neither Bayer Healthcare nor the Natural Source Vitamin E Association provided any input into the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Of relevance here as ivermectin and other therapeutics also have significant antiviral and anti-inflammatory properties and several patients had significantly elevated creatine levels, a marker for myoglobin, further complicating treatments.
COVID infection or molecular mimicry of spike protein created by transfection may lead to either direct or autoimmune damage to striated muscle fibres by macrophages and inflammatory cytokines.
The nephrotoxic breakdown products of this highly inflammatory process, such as the shorter lived myoglobin can then enter circulation and precipitate in renal tubules, leading to acute kidney injury (AKI) if left untreated.
Any medicines under consideration for administration need to be carefully considered if the patient has loss of kidney function:
Use of medicines for covid-19 treatment in patients with loss of kidney function: a narrative review (2021)
Covid-19 has been identified as the cause of acute respiratory disease with interstitial and alveolar pneumonia, but it can affect several organs, such as kidneys, heart, blood, nervous system and digestive tract. The disease-causing agent (Sars-CoV-2) has a binding structure to the angiotensin-converting enzyme 2 (ACE2) receptor, enabling entry into cells that express ACE2, such as the pulmonary alveolar epithelial cells. However, studies also indicate the possibility of damage to renal cells, since these cells express high levels of ACE2. Currently, there is no evidence to indicate a specific treatment for covid-19. Several drugs have been used, and some of them may have their excretion process altered in patients with abnormal kidney function. To date, there are no studies that assist health professionals in adjusting the dose of these drugs. Thus, this study aims to review and discuss the topic, taking into account factors associated with kidney injury in covid-19, as well as pharmacokinetic aspects and dose recommendations of the main drugs used for covid-19.
An in vitro study with renal proximal tubular epithelial cells established that Sars-CoV manifested persistent and productive infection, which was partially correlated with ACE2.6 expression using state-of-the-art single-cell techniques, Zou and colleagues showed high stratified organs and low risk, according to the level of ACE2 expression, indicating the kidneys as high-risk organs.7 These findings indicate the possibility of Sars-CoV-2 infecting renal cells.
On the other hand, drugs with potential efficacy against covid-19 have been increasingly studied and used in diagnosed patients, especially in the hospital environment.8 We know that the metabolism and clearance of many drugs depend on normal kidney function. These drugs can be altered by various pharmacokinetic processes in situations of kidney function deficit, subsequently resulting in adverse effects and clinical signs of drug intoxication in these patients.9 , 10
To date, there are no studies that assist health professionals in adjusting the dose of drugs used to treat covid-19 in patients with loss of kidney function. Thus, the aim of this study is to discuss the complexity of the topic, taking into account factors associated with the pathophysiology of acute kidney injury (AKI) and kidney abnormalities in covid-19, as well as pharmacokinetic aspects and recommendations for doses of the main drugs used for covid-19.
Legends: APD/CAPD (automated peritoneal dialysis/continuous ambulatory peritoneal dialysis); bid (twice a day); CAV/VVHD (continuous arteriovenous hemofiltration/veno-venous hemofiltration); D (dialyzed); GFR (glomerular filtration rate); HD (intermittent hemodialysis); HDF/AF (intermittent hemodialysis/high flow); PO (oral administration); I (improbable); IV (intravenous administration); L (lopinavir); ND (not dialyzed); NR (not reported); P (prophylaxis); R (ritonavir); SQ (subcutaneous administration); T (treatment).
*Body Mass Index (BMI) ≥ 40 kg/m2: increase the dose in about 30%; or 0.5 mg/kg/day using current weight for the calculation.
About 50% of chloroquine is excreted unchanged, and 10% as a metabolite by the kidneys, which can lead to its buildup in the body, in addition to further prolonging the drug's half-life, which is already high (10-60 days).18 Therefore, a 50% dose reduction is recommended in patients with GFR < 10 mL/min. Regarding patients receiving dialysis, there is no recommendation to supplement the dose for most existing dialyses.8 , 18 Hydroxychloroquine is only 3% excreted unchanged, but it is metabolized to chloroquine and active metabolites that can also buildup in patients with kidney injury. Thus, a 50% reduction is recommended in patients with GFR < 30 mL/min. There is no need to supplement the dose of these drugs for any of the existing dialysis treatments.18
AZITHROMYCIN
There is no recommendation to adjust the dose of azithromycin in patients with abnormal kidney function. However, it is important to use it with caution in patients with GFR < 10 mL/min, as it can increase adverse gastrointestinal effects, such as diarrhea, nausea and vomiting due to the increase (around 33%) of the systemic exposure of azithromycin.15 , 18 Furthermore, as azithromycin is associated with hydroxychloroquine in the treatment of covid-19, and both may be accumulating in the body of these patients, effective electrocardiogram monitoring must be performed due to the greater risk of QT interval prolongation.15 , 16 , 17 It is recommended that azithromycin be administered within 4 hours of hydroxychloroquine. There is no need to supplement the dose for any of the existing dialysis treatments.18
ANTIPARASITIC
There are no dose adjustments provided by the manufacturer for nitazoxanide. However, this drug is excreted unchanged by the kidneys by up to 33%, and can be accumulated in the body of patients with abnormal kidney function, as well as its active metabolite (tizoxanide), which is highly linked to plasma proteins (> 99%), being able to be displaced, raising the free fractions in these patients.15 In addition, a study carried out in rats showed a significant increase in the serum level of creatinine and urea one day after treatment with nitazoxanide, when compared with the control group.36 it results in the use of this medication in patients with abnormal kidney function.17 There are no studies that support the need for dose supplementation in any of the dialysis treatments used.17 , 18
There are also no dose adjustments provided by the manufacturer for Ivermectin. However, this drug is highly linked to plasma proteins (93%), mainly albumin, and can be displaced, increasing free fractions in patients with loss of capacity.15 , 16 In addition, a study carried out in rats showed that the use of Ivermectin can compromise kidney and liver integrity.37 There are no studies that support the need for dose supplementation for any of the existing dialysis treatments.17 , 18
CORTICOSTEROIDS
There is no recommendation to adjust the dose in patients with abnormal kidney function, although 65% of dexamethasone is excreted unchanged within 24 hours. However, the use of corticosteroids should be done with caution in patients with loss of kidney function, as greater fluid retention may occur. Dexamethasone should also be used with caution in the elderly, always in the lowest possible dose.15 , 16 There is no need to supplement the dexamethasone dose for any of the existing dialysis treatments. On the other hand, patients using methylprednisolone should receive the usual doses after dialysis treatment.18
TOCILIZUMAB
There is no recommendation for dose adjustment in patients with abnormal kidney function. However, this drug has a high molecular weight (148 kDa), and it is unlikely to be significantly eliminated by the renal route in patients with GFR < 30 mL/min. Thus, we recommend caution regarding its use and monitoring of kidney function in these patients.15 , 16 There are no studies that support the need for dose supplementation for any of the existing dialysis treatments.18
ANTICOAGULANTS
There is no recommendation to adjust the dose of unfractionated heparin in patients with abnormal kidney function. However, there may be greater renal elimination (around 50%) at high doses, in addition to an increase in the drug's half-life.15 , 16 , 18 Therefore, caution should be exercised when using it in high doses. There is no need to supplement the dose for any of the existing dialysis treatments.18
In contrast, enoxaparin needs dose adjustment in these patients, since there is a risk of bleeding due to decreased renal clearance (about 30%) and increased bioavailability. In addition, although 10% of the drug is excreted unchanged by the kidney, a large part of the active and inactive metabolites (around 40%) is excreted by this route and can accumulate in these patients. Finally, 80% of the drug is bound to plasma proteins, being able to be displaced and increasing free fractions in patients with loss of kidney function.15 , 16 Therefore, a dose reduction to 20 to 30 mg/day is recommended (prophylactic ) or 0.5-1 mg/kg/day (treatment) in patients with GFR < 30 mL/min.15 , 38 Regarding the existing dialysis treatments, there is no need for dose supplementation, except for intermittent hemodiafiltration, in which it is recommended to administer an additional dose similar to the dose used in patients with GFR < 30 mL/min.18
ANTIVIRALS
There are no dose adjustments for lopinavir/ritonavir provided by the manufacturer, and a reduction in the clearance of these drugs is unlikely in patients with kidney injury. However, these drugs are highly bound to plasma proteins (> 98%) and can be displaced, increasing free fractions in these patients.15 , 16 It should also be used with caution in elderly patients, since the data on these patients are insufficient to determine whether they respond differently from adults.39 , 40 , 41 There is no need for dose supplementation for any of the existing dialysis treatments.18
Oseltamivir is a prodrug, extensively metabolized by esterases in the liver to the active carboxylate metabolite. This metabolite is eliminated by renal excretion (99%). Furthermore, renal clearance exceeds the glomerular filtration rate, indicating that tubular secretion occurs in addition to glomerular filtration.15 , 18 This metabolite can accumulate in the body of patients with abnormal kidney function. Thus, a dose of 30 mg twice daily is recommended for patients with GFR between 31 and 60 mL/min; 30 mg daily to patients with GFR between 10 and 30 mL/min; and 30 mg single dose to patients with GFR < 10 mL/min.17 , 18 In patients undergoing hemodialysis and peritoneal dialysis, an initial dose of 30 mg can be administered before starting dialysis and supplemented with 30 mg each session or 5 days, respectively.18 Continuous or venous arteriovenous hemofiltration is similar to hemodialysis. For intermittent hemodiafiltration, the doses indicated will depend on the flow rate, which is detailed at the bottom of Table 2.18
For favipiravir, there are no dose adjustments provided by the manufacturer, because available data is limited.17 However, this drug undergoes hepatic metabolism, generating metabolites that are excreted in renal hydroxylated forms. The fraction of metabolites excreted in the urine increases over time, reaching 80-100% after 7 days.42 Thus, it is important to monitor patients with loss of kidney function, as well as the elderly. There are no studies that support the need for dose supplementation for any of the existing dialysis treatments.17
Finally, there are no studies recommending dose adjustment of remdesivir in patients with loss of kidney function, as there are no safety or pharmacokinetic data available for this population. However, animal studies have shown an increase in urea and mean creatinine, as well as renal tubular atrophy in histopathological findings, indicating altered kidney function;43 therefore, use in patients with GFR < 30 mL/min is not recommended.8 Patients receiving Kidney Replacement Therapy (KRT) were excluded from current clinical trials.15
CONCLUSION
The topics presented in this study show that patients who have abnormal kidney function and were affected by covid-19 undergo several physiological changes that can cause changes in the pharmacokinetics and pharmacodynamics of drugs, which can cause variations in their serum concentrations and, consequently, risk of overdosing and toxicity.
In addition, many drugs used to treat covid-19 require dose adjustments or continuous monitoring in this population.
Thus, the indication and use of these drugs must be well evaluated, checking the risk-benefit of the therapy, taking into account the particularities of each patient.
Nunes LLA, Lima TM. Use of medicines for covid-19 treatment in patients with loss of kidney function: a narrative review. J Bras Nefrol. 2021 Apr-Jun;43(2):254-262. doi: 10.1590/2175-8239-JBN-2020-0105. PMID: 33316027; PMCID: PMC8257283.
From COVID-19 vaccine induced rhabdomyolysis: Case report with literature review (2021):
1. Introduction
Coronavirus Disease 2019 (COVID-19) caused a significant impact on the health, economic and political systems in 2020, and by the end of the year, hope was born with the introduction of COVID-19 vaccines aiming at ending the pandemic. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, is an enveloped, positive-sense, single-stranded RNA virus with viral spike glycoproteins, and the studied mechanism has contributed significantly to vaccination efforts and public health initiatives [1]. The coronavirus spike protein has been shown to mediate membrane fusion via the binding of cellular receptors [2]. Herein we present the first case of COVID-19 vaccine-induced rhabdomyolysis to help clinicians easily identify such a problem in newly vaccinated patients.
1.1. Case presentation
We present a 21-year-old male patient with a past medical history of asthma who presented to the emergency department for progressively worsening pain and swelling in the lower back for one day after his first Pfizer/BioNTech COVID-19 vaccine injection. He described it as a 5 to 10 out of 10 sharp pain located at his mid to lower back with radiation to his left lateral thigh. The pain worsened with body movement. The patient tried over-the-counter pain medication with limited relief. He also noticed a darkened urine color before he came to the hospital.
The patient did not use any medication regularly. He denied excessive exercise, heavy weightlifting or body trauma after vaccination. He had no family history of autoimmune or musculoskeletal diseases, and surgical history was only significant for an uncomplicated appendectomy. Patient endorsed social marijuana use but denied other drug, alcohol, or tobacco use.
Transient elevated blood pressure was noticed at the beginning of the hospitalization, but other vital signs were unremarkable. Physical examination was positive for tenderness to the paraspinal lumbar area upon palpation. The straight leg test was negative.
Pertinent lab results included Creatinine Phosphokinase (CPK) level more than 22,000 U/L (normal range 20–190 U/L), Aldolase 97.8 U/L (normal range 3.3–10.3 U/L), alanine aminotransferase 165 U/L (normal range 0–41 U/L), aspartate aminotransferase 675 U/L (normal range 5–40 U/L), high sensitive C-reactive protein 6.4 mg/L (normal range < 5.0 mg/L) and Lactate dehydrogenase 1525 U/L (normal range 135–225U/L). Urinalysis revealed clear yellow urine with positivity for blood and protein but negativity for Red Blood Cells (RBCs). Basic Metabolic Panel (BMP) showed normal potassium, bicarbonate, Blood Urea Nitrogen, and creatinine levels. Hepatitis B and C panels were negative except for hepatitis B surface antigen. Total and free carnitine levels were within the normal range. SARS-CoV-2 PCR was negative.
Anti-aminoacyl-tRNA synthetase antibodies (ab) (includes anti-Jo-1, anti-PL-7, anti-PL-12, anti-EJ, anti-OJ), anti-SRP ab, anti-MI-2 ab, anti-TIF1-gamma ab, anti-MDA5 ab, anti-NXP2 ab, anti-PM/Scl-100 ab, anti-U3 RNP ab, U2 snRNP Ab, ANTI-U1-RNP AB, anti-Ku antibodies, anti–SS–A 52 KD ab IgG, ANA, and HIV antibodies were all negative. ESR, haptoglobin, TSH, and cortisol were in the normal range.
No musculoskeletal abnormalities were found on CT scan of the thoracic and lumbar spine. The right upper quadrant sonogram showed no abnormality in the liver, biliary tracts, gallbladder, or pancreas.
The patient had been hydrated with high volume IV normal saline for rhabdomyolysis. Morphine was given as needed for muscle pain. Follow-up lab results showed down-trending CPK and AST levels. Electrolyte level and renal functions have been within normal limits throughout the hospitalization. No change of urine color or urine output volume was noticed. The patient felt pain improved after treatments and was discharged after five days of hospital stay.
The classic triad on presentation includes myalgia, weakness, and dark urine. Other constitutional signs such as fever, chills, malaise, nausea, vomiting, tachycardia, muscle swelling, and tenderness can occur but are non-specific. A thorough history and physical examination are paramount to diagnosing rhabdomyolysis, considering the classic triad is present in less than 10% of patients. Patients with known risk factors (trauma, exertion, drugs, muscular disease, infection) and suggestive physical and laboratory findings should be suspected for this syndrome, and plasma creatine kinase (CK) determination should be done. Creatine kinase has been widely accepted as the gold standard over myoglobin, due in part to its long half-life of 36 h (vs. 2–4 h of myoglobin), thus decreasing false-negative results. In addition, urine myoglobin can be high, and muscle biopsy can confirm the diagnosis, although it is rarely needed [13].
Most clinicians use an arbitrary CK value of five times the upper limit of normal to guide therapy. However, its use in predicting the risk of acute kidney injury has not been formally established. Other clinical factors should be used to assess disease severity, such as volume status, urine output, concurrent sepsis or organ failure, electrolyte homeostasis, or comorbid conditions. Regardless of the cause of injury, myoglobin and muscle enzymes (creatine kinase, aldolase, lactate dehydrogenase) and electrolytes leak into the extracellular space leading to the complications seen in this syndrome. Myoglobin is nephrotoxic and can precipitate in the renal tubules leading to acute kidney injury, the most serious complication which can occur in up to one-third of patients [14]. Hyperkalemia and hyperphosphatemia can occur through direct intracellular release, and rapid accumulation of potassium in the serum can lead to malignant ventricular arrhythmias and cardiac arrest. Calcium salts can deposit in the damaged muscle causing severe hypocalcemia. Elevated liver enzymes can be seen, and isolated high aspartate aminotransferase (with a normal alanine aminotransferase) could be a clue that rhabdomyolysis is occurring. A global hypovolemic state can ensue as intravascular fluid is third spaced and remains sequestered into injured muscles, further increasing the risk of prerenal acute kidney injury, and in some instances, even causing a compartment syndrome. The release of prothrombotic substances from damaged muscles can activate the clotting cascade and lead to disseminated intravascular coagulation.
1.5. COVID and rhabdomyolysis
Rhabdomyolysis causes muscle cell injury and breakdown. The mechanism behind SARS-CoV2 causing rhabdomyolysis is not clear. However, viral cell invasion and cytokine-mediated direct muscle cell damage have been implicated [19]. Notably, Type II pneumocyte cytoplasm in early stages and colonic mucosa in COVID-19 patients presented with diarrhea contained the viral particles [19]. In the related MERS-CoV (Middle East Respiratory Syndrome) cases from 2005, viral particles were found in the skeletal muscle-invading macrophages [20]. Since both SARS-CoV2 and the related MERS-CoV use the angiotensin-converting enzyme II receptors, this is a highly suggested route for SARS-CoV2 viral invasion [20]. ACE2 receptors are found in several organs, including oral and nasal mucosa, lungs, small intestine, colon, liver, and kidneys [21].
…Unfortunately, rhabdomyolysis with its symptoms such as fever, myalgia, and signs (elevated liver enzymes and lactate dehydrogenase) is similar to COVID-19 itself and may go unrecognized [22]. This is concerning as COVID-19 patients with concomitant rhabdomyolysis were found to have an increased risk of deterioration, increased risk of ICU admission (90.9% vs. 5.3%), increased mechanical ventilation (86.4% vs. 2.7%), and increased risk of in-hospital death [28]. Two independent risk factors for in-hospital deaths were creatine kinase (CK) levels greater than 1000 IU/L and serum myoglobin levels greater than 1000 ng/ml [28]. However, Bach et al. clarify that while both CK and myoglobin levels are monitored, Myoglobin levels are less useful in diagnosing rhabdomyolysis due to increased false-negative results, caused by myoglobin's short half-life [23]. However, this suggests the need for CK monitoring in all SARS-CoV2 patients to screen for rhabdomyolysis.
Nassar M, Chung H, Dhayaparan Y, Nyein A, Acevedo BJ, Chicos C, Zheng D, Barras M, Mohamed M, Alfishawy M, Nso N, Rizzo V, Kimball E. COVID-19 vaccine induced rhabdomyolysis: Case report with literature review. Diabetes Metab Syndr. 2021 Jul-Aug;15(4):102170. doi: 10.1016/j.dsx.2021.06.007. Epub 2021 Jun 15. PMID: 34186348; PMCID: PMC8205294.
Clinicopathological Characteristics of Inflammatory Myositis Induced by COVID-19 Vaccine (Pfizer-BioNTech BNT162b2): A Case Report (2022)
Abstract
As more individuals were coronavirus disease 2019 (COVID-19) vaccinated, unexpected side effects appeared. Herein, we present the case of a 30-year-old man with myopathy in both extremities after the second dose of the Pfizer-BioNTech (BNT162b2) COVID-19 vaccine. Symptoms, swelling and pain, started from the proximal upper and lower extremities and extended to the distal parts. Although he underwent massive hydration, the muscle enzyme level continuously increased. He complained of dysphagia and dysarthria. Microscopically, muscle biopsy showed multifocal or scattered macrophage infiltration and degenerated myofibers. In contrast to general myopathy including inflammatory myositis and rhabdomyolysis, vaccine-induced inflammatory myositis shows a prolonged increase in muscle enzyme levels and multifocal macrophage infiltration with necrosis of the muscle fibers. Symptoms improved with glucocorticoid and immunosuppressive treatment. If vaccinated individuals experience severe and continuous muscle pain and swelling, clinicians should consider vaccine-induced inflammatory myositis, measure the muscle enzyme levels, and perform muscle biopsy for a definite diagnosis.
…Microscopically, muscle biopsy showed multifocal or scattered macrophage infiltration and degenerated myofibers. In contrast to general myopathy including inflammatory myositis and rhabdomyolysis, vaccine-induced inflammatory myositis shows a prolonged increase in muscle enzyme levels and multifocal macrophage infiltration with necrosis of the muscle fibers. Symptoms improved with glucocorticoid and immunosuppressive treatment. If vaccinated individuals experience severe and continuous muscle pain and swelling, clinicians should consider vaccine-induced inflammatory myositis, measure the muscle enzyme levels, and perform muscle biopsy for a definite diagnosis.
Erythematous purpuric plaques on the trunk (A) and both hands (B).
Kim JH, Kim JH, Woo CG. Clinicopathological Characteristics of Inflammatory Myositis Induced by COVID-19 Vaccine (Pfizer-BioNTech BNT162b2): A Case Report. J Korean Med Sci. 2022 Mar 21;37(11):e91. doi: 10.3346/jkms.2022.37.e91. PMID: 35315602; PMCID: PMC8938612.
There is a correlation between the presence of parasites and cancer occurrence. Some primary effects are indicated but in the main causation appears to be mainly secondary and opportunistic in nature according to studies. And the importance of maintaining a healthy gut biome cannot be understated.
As with other co-infections in the cancer patient they should certainly be treated as they can be pro-inflammatory which promotes tumorigenesis and may contribute to the suppression of immune response due to exhaustion and general malaise, in a feedback loop.
The International Agency for Research on Cancer has estimated that 16% of cancer worldwide is caused by infectious factors, including parasites.
Colorectal cancer and Blastocystis sp. infection (2021)
Abstract
Background
Blastocystis sp. is a common intestinal protozoan found worldwide. Based on gene analysis, 17 subtypes (STs, ST1–ST17) have been identified, 9 of which have been isolated from humans. Differences in clinical consequences may depend on differences among the STs. Here, we evaluated the prevalence of Blastocystis sp. in patients with colorectal cancer (CRC) compared to a control group and assessed the relationships between Blastocystis sp. infection and sex; age; and CRC grade, stage, and location.
Methods
The study included 107 CRC patients (41 women and 66 men, median age 65 years); 124 subjects without colorectal cancer or a history of oncological disease comprised the control group (55 women and 69 men, median age 63). Stool samples were collected from patients before oncological treatment and examined using light microscopy (iodine-stained smear). Additionally, PCR-based identification of Blastocystis sp. was performed in 95 stool samples from CRC patients and 76 stool samples from the control group.
Results
Light microscopy showed that the prevalence of Blastocystis sp. was significantly higher in CRC patients than in the control group (12.15% and 2.42%, respectively; p = 0.0041). Multivariate analysis showed that the odds of Blastocystis sp. infection were fivefold higher in the CRC group than in the control group. PCR-based molecular examinations demonstrated that the proportion of patients infected with Blastocystis sp. was significantly higher in the CRC group than in the control group (12.63% and 2.63%, respectively; p = 0.023). The predominant ST in the CRC group was ST3, detected in nine patients (75%), followed by ST1 (2 patients, 16.7%) and ST2 (1 patient, 8.3%). No association was found between Blastocystis sp. infection and age, sex, or CRC stage, grade, or location.
Conclusions
The results showed that CRC was associated with an increased risk of opportunistic Blastocystis sp. infection, even before oncological treatment. To the best of our knowledge, this is the first report estimating the prevalence of Blastocystis sp. infection in CRC patients before oncological treatment in Europe.
Background
The International Agency for Research on Cancer has estimated that 16% of cancer worldwide is caused by infectious factors, including parasites. Colorectal cancer (CRC) is one of the most common neoplasms in humans. Most CRCs are sporadic, and the contributions of environmental risk factors have been widely investigated [2]. Microbes colonizing the gut are also considered potential cancer risk factors [3, 4].
Blastocystis sp. is a common parasitic protozoan with a worldwide distribution that is found in the gastrointestinal (GI) tract of humans and a wide range of animal hosts [5, 6]. Its prevalence in humans is estimated to be as high as 10% in developed countries and 50–60% in developing countries [7]. Blastocystis sp. is transmitted through the faecal–oral route as well as through contaminated water and food [8, 9]. The pathogenicity of this protozoan is controversial, as it causes non-specific digestive tract symptoms, such as abdominal pain, nausea, vomiting, anorexia, acute or chronic diarrhoea, and weight loss. Blastocystis sp. infection is usually associated with alternating episodes of diarrhoea, normal defecation, and even constipation [10]. Poirier et al. suggested an association between Blastocystis sp. infection and irritable bowel syndrome (IBS) [11]. On the other hand, a higher rate of Blastocystis sp. infection in asymptomatic patients than in those with IBS symptoms was detected in Denmark [12].
The clinical significance of Blastocystis sp. infection remains uncertain, mainly because of its common occurrence in both dyspeptic patients and healthy individuals [9, 13]. Some studies have maintained that Blastocystis sp. is part of a healthy gut microbiome [13, 14]. However, it has also been reported that Blastocystis sp. infection can have features of opportunistic infection, as has been observed in patients with CRC treated with chemotherapy [15].
The identification of this organism at the species level is difficult. Blastocystis was originally named B. hominis, but subsequent phylogenetic studies limited the name to “Blastocystis species” because of the genetic diversity among members within the genus [16]. It was discovered that host specificity and the pathogenic potential of different isolates are correlated with sequence variations in the small subunit ribosomal RNA (SSU-rRNA) gene [17]. Based on these variations, members of the genus have been ordered into several subtypes (STs) [18]. Based on SSU-rRNA gene analysis, 17 STs (ST1–ST17) have been identified, 9 of which have been isolated from humans [19]. Differences in clinical consequences may depend on differences among STs [20]. Because Blastocystis sp. are found in both symptomatic and asymptomatic patients, the pathogenicity of this organism remains unclear [21,22,23]. Some studies have shown an association between Blastocystis sp. ST variation and pathogenicity. Dogruman-Al et al. [24] suggested that ST2 is a non-pathogenic genotype of Blastocystis sp.
The predominance of Blastocystis sp. ST3 among patients with chronic GI illness has been shown in Malaysia [25], Singapore [26], Egypt [27], Turkey [28], the United States [29], and Iran [30]. Khademvatan et al. [20] showed that in southern Iran, the most common ST of Blastocystis sp. was ST3, which correlated with the presence of GI symptoms in 44.83% of cases.
Studies evaluating the prevalence of Blastocystis sp. in the French population (inhomogeneous population of 788 patients from 11 hospitals) showed that the frequency of Blastocystis sp. infection in patients with symptoms of GI disorders was not significantly higher than that in patients without symptoms, and the most common ST was ST3 [31].
The aim of this study was to evaluate the prevalence of Blastocystis sp. in patients with CRC compared to that in a control group without colorectal cancer or a history of oncological disease and to assess the relationship of Blastocystis sp. infection with the sex and age of the subjects as well as CRC stage, grade, and location. To the best of our knowledge, this is the first report estimating the prevalence of Blastocystis sp. infection in CRC patients before oncological treatment in Europe.
Discussion
Microbiota alterations, referred to as dysbiosis, are often associated with CRC. Both human studies and studies conducted in animals showed that the gut microbiota related to CRC was distinct from that in subjects without CRC [35,36,37]. In addition, two meta-analyses of faecal metagenome changes specific to CRC were published in 2019 [38, 39]. The human gut microbiota comprises bacteria, viruses, and eukaryotes (e.g., protozoa, helminths, and fungi). In human CRC samples, cytomegalovirus (CMV), John Cunningham (JC) virus, and human papillomavirus (HPV) have been identified, although the data are conflicting [37, 40]. Additionally, changes in the mycobiome associated with human CRC have been reported [41].
The mechanism of the impact of dysbiosis on CRC carcinogenesis encompasses inflammation, immune regulation, metabolism of dietary components, and genotoxin production [42]. The gut microbiota interacting with the host immune system can affect the inflammatory process in the GI tract [43]. The microbiota produce numerous metabolites significant for human physiology [44]; on the other hand, these metabolites can impact the risk of developing CRC. Another carcinogenic mechanism of the microbiota is the production of DNA-damaging toxins [40, 45, 46]. In a driver–passenger model of CRC, the mucosa of the colon is colonized by pathogenic driver bacteria, which produce genotoxins that induce inflammation and, consequently, the adenoma-carcinoma sequence [47]. On the other hand, opportunistic passenger bacteria might proliferate in CRC tumours and stimulate the infiltration of immune cells [40, 47].
Disruptions in the gut microbiota and changes in its relative abundance can alter the balance, leading to many diseases, including inflammatory bowel disease (IBD) and Clostridium difficile infection [48, 49]. Impairment of the symbiotic relationship between the microbiota and the host leads to immune dysregulation and can induce chronic inflammation, resulting in IBD [48]. The microbiota composition varies between certain subtypes of IBD (Crohn’s disease, colitis ulcerosa) and the presence of an active phase [50,51,52]. The role of gut microbiota alterations in IBD has been widely examined [53, 54]. However, in our study, we focused on patients with CRC, and none of our patients suffered from IBD.
The pathogenic potential of Blastocystis sp. remains controversial [55,56,57]. Blastocystis sp. interact with bacterial gut microbes [57, 58]. The increased prevalence of Blastocystis sp. is related to changes in the composition of the microbiota in the human host [59,60,61,62]. Modifications of the microbiota affect the host immune response [55, 63].
To the best of our knowledge, this is the first report to estimate the prevalence of Blastocystis sp. infection in CRC patients before oncological treatment in Europe. Few studies have examined Blastocystis sp. infection in CRC patients (those that have were from Uzbekistan, Saudi Arabia, Turkey, and Malaysia) [64,65,66,67]. Most studies indicate opportunistic characteristics of Blastocystis sp. infection, but there are also reports indicating that Blastocystis sp. is a component of the healthy gut microbiome [14].
Our results were obtained from a homogeneous group of patients with CRC before oncological treatment, and individuals in the control group were matched by age to the CRC patients. The results showed that the odds of Blastocystis sp. infection patients with CRC were 5 times higher than those in the control group; these results were obtained not only by LM but also by PCR, and the proportion of individuals infected with Blastocystis sp. was significantly higher in the CRC group (12.63%) than in the control group (2.63%). Chandramathi et al. [15] demonstrated the opportunistic characteristics of Blastocystis sp. infection among patients with CRC (n = 15). Our results suggest that CRC is related to an increased risk of opportunistic infection with Blastocystis sp. even before oncological treatment, which may have additional effects on the immunological system.
At the time of planning of our study, we could not predict what the results would be, because Blastocystis sp. can be considered a marker of a healthy gut microbiota [57]; however, it is not possible to exclude opportunistic Blastocystis sp. infection in patients with CRC.
A significantly higher prevalence of Blastocystis sp. (80%) was found using LM in 200 CRC patients compared to the control population in Tashkent, Uzbekistan [64]. The prevalence of Blastocystis sp. in CRC patients was fourfold higher than that in the control population. However, the authors did not conduct PCR analysis, which could have enabled the determination of the ST of Blastocystis sp. [64]. Yersal et al. [66] detected Blastocystis sp. in 6.5% of 232 stool samples from cancer patients suffering from different types of cancer (lung, breast, CRC) by LM, but in the CRC patients, Blastocystis sp. was detected using PCR in 7.5% of the 66 examined cases [66], among which ST1 was the predominant ST (3 cases, 60%), followed by ST3 (2 cases, 40%). In our study, ST3 was the predominant ST in CRC patients (9 patients, 75%), followed by ST1 (2 patients, 16.7%) and ST2 (1 patient, 8.3%).
Kumarasamy et al. found Blastocystis sp. infection prevalence rates of 22.08% in 204 Malaysian CRC patients and 9.95% in the control group [67]. The most common ST was ST3 (12.75%), followed by ST1 (4.41%), ST2 (0.49%), and ST5 (0.49%) [67].
Blastocystis sp. infection was confirmed in 74 CRC patients (29.7%) in Saudi Arabia [65], where ST1 was the most predominantly detected ST (54,5%) with a significant risk association (crude OR: 7.548; 95% CI 1.629–34.987; p = 0.004).
On the other hand, Beghini et al. [14] showed a lower frequency of Blastocystis sp. infection among patients with CRC (5.7%) compared to the healthy control group in their analysis of 12 studies of patient populations from different continents (n = 1689) with different diseases of the GI tract, including CRC. It is worth noting that only 53 of those patients suffered from CRC [3, 14].
Differences in the results of studies on the prevalence of various Blastocystis sp. STs may be related to the genetic diversity of Blastocystis sp., non-homogeneity of the analysed patient groups, and ethnic diversity among the examined populations inhabiting different parts of the world [56].
A possible role for Blastocystis sp. in CRC pathogenesis has been suggested [15, 64, 68, 69]. Postulated potential carcinogenic effects of Blastocystis sp. infection in humans, especially CRC patients, was examined by Chan et al. [70], and the ability of Blastocystis sp. to induce the growth of CRC cell lines by inhibiting the apoptotic effects of CRC cells has been documented [70]. Furthermore, isolated antigens of Blastocystis sp. isolates were shown to promote the proliferation of cancer cells via downregulation of host immune cellular responses [22, 68, 70]. Chan et al. [70] showed that antigens isolated from symptomatic human hosts caused a more extensive inflammatory reaction and a higher proliferation rate of CRC cells than isolates from asymptomatic human hosts. Chandramathi et al. [68] also showed that solubilized antigen of Blastocystis sp. facilitates growth in the human HCT116 CRC cell line. The antigen isolated from ST3 had the most prominent effect on the proliferation of CRC cells [71], which confirmed the case of severe ST3 Blastocystis infection in a 35-year-old man at the time of CRC diagnosis, as described by Padukone et al. [72].
The possible impact of Blastocystis sp. infections on CRC carcinogenesis remains unclear. The pathogenicity of Blastocystis sp. is suspected to be caused by the release of cysteine proteases by this protozoan. These proteases stimulate mucosal cells to release interleukin 8, which has been associated with gut inflammation [56].
Chronic inflammation is an established risk factor for CRC [73]. Among the molecular mechanisms of CRC pathogenesis, oxidative stress plays an important role and has been shown to be associated with Blastocystis sp. infection [69].
In our study, Blastocystis sp. infection was confirmed by both LM and PCR in all nine samples positive for ST3 from patients with CRC, but no such confirmation was found for the ST1 or ST2 genotypes. In three CRC patients, Blastocystis sp. infection detected by LM was not confirmed by PCR, possibly due to the presence of other organisms in the stool samples and non-specific amplification, which was revealed by sequencing of the obtained PCR products. A possible explanation for this discrepancy could also be a misdiagnosis of the parasite by light microscopy.
However, there was a strong positive correlation between the LM and PCR results in the CRC group (phi coefficient = +0.71). The presented results show that LM is an inexpensive, sensitive, and accessible method in daily practice. On the other hand, the PCR method allows for the identification of STs that may have different pathogenic potentials.
Conclusions
We demonstrated that the prevalence of Blastocystis sp. was higher in CRC patients than in the control group, independent of the diagnostic method used. In addition, ST3 was the predominant Blastocystis sp. ST among Polish CRC patients. Furthermore, Blastocystis sp. infection occurred five times more often in the CRC group than in the control group, independent of age, sex, and diagnostic method. No association was found between Blastocystis sp. infection and age, sex, staging, grading, or CRC location. Together, these results show that CRC is associated with an increased risk of opportunistic Blastocystis sp. infection even before oncological treatment. The potential relationship of Blastocystis sp. with CRC carcinogenesis needs further study (Additional file 1).
Sulżyc-Bielicka, V., Kołodziejczyk, L., Adamska, M. et al.Colorectal cancer and Blastocystis sp. infection. Parasites Vectors 14, 200 (2021). https://doi.org/10.1186/s13071-021-04681-x
It is certainly a significant secondary therapeutic benefit that many therapeutics with anti-tumor properties are also anti-parasitic in nature or first administered for that (hydroxychloroquine, ivermectin, artemisin). This is no coincidence as some of the modes of inhibition overlap.
In contrast, either familial or acquired mutations in tumor suppressor genes such as TP53 or KRAS are a factor in up to 90% of some cancer types:
From Gain-of-Function Mutations in the Tumor Suppressor Gene p53 (2000):
Abstract
The tumor suppressor protein p53 is a multifunctional transcription factor involved in the control of cell cycle progression, DNA integrity, and cell survival. p53 is mutated in half of all tumors and has a wide spectrum of mutation types. p53 mutants show different degrees of dominance over coexpressed wild-type p53, and loss of the wild-type p53 allele has been observed frequently. Several p53 mutants can exert oncogenic functions beyond their negative domination over the wild-type p53 tumor suppressor functions. These so-called gain-of-function effects, such as enhancement of tumorigenicity and therapy resistance, were investigated in p53-null cells. The possible mechanisms by which p53 mutants exert their gain-of-function effects are reviewed. The existence of functional gains of certain p53 mutants has important ramifications for tumor prognosis and cancer therapies.
The tumor suppressor gene p53 is mutated in 50% of all tumors, and it plays a role in the carcinogenesis of many different malignancies. The gene is mutated in more than 90% of head and neck squamous cell carcinomas. In contrast, the incidence of p53 mutations is very low in hematological malignancies.
Monique G. C. T. van Oijen, Pieter J. Slootweg; Gain-of-Function Mutations in the Tumor Suppressor Gene p53 . Clin Cancer Res 1 June 2000; 6 (6): 2138–2145.
The KRAS gene provides instructions for making a protein called K-Ras that is part of a signaling pathway known as the RAS/MAPK pathway. The protein relays signals from outside the cell to the cell's nucleus. These signals instruct the cell to grow and divide (proliferate) or to mature and take on specialized functions (differentiate). The K-Ras protein is a GTPase, which means it converts a molecule called GTP into another molecule called GDP. In this way the K-Ras protein acts like a switch that is turned on and off by the GTP and GDP molecules. To transmit signals, it must be turned on by attaching (binding) to a molecule of GTP. The K-Ras protein is turned off (inactivated) when it converts the GTP to GDP. When the protein is bound to GDP, it does not relay signals to the cell's nucleus.
The KRAS gene belongs to a class of genes known as oncogenes. When mutated, oncogenes have the potential to cause normal cells to become cancerous. The KRAS gene is in the Ras family of oncogenes, which also includes two other genes: HRAS and NRAS. These proteins play important roles in cell division, cell differentiation, and the self-destruction of cells (apoptosis).
Lung cancer
At least three mutations in the KRAS gene have been associated with lung cancer. Lung cancer is a disease in which certain cells in the lungs become abnormal and multiply uncontrollably to form a tumor. Lung cancer may not cause signs or symptoms in its early stages. These KRAS gene mutations are somatic, which means they are acquired during a person's lifetime and are present only in tumor cells. Somatic mutations are not inherited. Nearly all of the KRAS gene mutations associated with lung cancer change the amino acid glycine at position 12 or 13 (Gly12 or Gly13) or change the amino acid glutamine at position 61 (Gln61) in the K-Ras protein. These mutations result in a K-Ras protein that is constantly turned on (constitutively activated) and directing cells to proliferate in an uncontrolled way, which leads to tumor formation. When these genetic changes occur in cells in the lungs, lung cancer can develop.
KRAS gene mutations are found in 15 to 25 percent of all lung cancer cases but are more frequent in white populations than in Asian populations; 25 to 50 percent of whites with lung cancer have KRAS gene mutations, whereas 5 to 15 percent of Asians with lung cancer have KRAS gene mutations.
KRAS gene mutations are much more common in long-term tobacco smokers with lung cancer than in nonsmokers. Lung cancers with KRAS gene mutations typically indicate a poor prognosis and are associated with resistance to several cancer treatments.
Other cancers
Somatic mutations in the KRAS gene are involved in the development of several types of cancer, particularly pancreatic and colorectal cancers. These mutations lead to a K-Ras protein that is more strongly overactivated than the mutations that cause cardiofaciocutaneous syndrome (described above). The abnormal K-Ras protein is always active and can direct cells to proliferate in an uncontrolled way.
From Ivermectin, ‘Wonder drug’ from Japan: the human use perspective (2011):
Ivermectin proved to be even more of a ‘Wonder drug’ in human health, improving the nutrition, general health and wellbeing of billions of people worldwide ever since it was first used to treat Onchocerciasis in humans in 1988. It proved ideal in many ways, being highly effective and broad-spectrum, safe, well tolerated and could be easily administered (a single, annual oral dose). It is used to treat a variety of internal nematode infections, including Onchocerciasis, Strongyloidiasis, Ascariasis, cutaneous larva migrans, filariases, Gnathostomiasis and Trichuriasis, as well as for oral treatment of ectoparasitic infections, such as Pediculosis (lice infestation) and scabies (mite infestation).14) Ivermectin is the essential mainstay of two global disease elimination campaigns that should soon rid the world of two of its most disfiguring and devastating diseases, Onchocerciasis and Lymphatic filariasis, which blight the lives of billions of the poor and disadvantaged throughout the tropics. It is likely that, throughout the next decade, well over 200 million people will be taking the drug annually or semi-annually, via innovative globally-coordinated Mass Drug Administration (MDA) programmes.
Crump A, Ōmura S. Ivermectin, 'wonder drug' from Japan: the human use perspective. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87(2):13-28. doi: 10.2183/pjab.87.13. PMID: 21321478; PMCID: PMC3043740.
Repositioning of Antiparasitic Drugs for Tumor Treatment (2021)
Abstract
Drug repositioning is a strategy for identifying new antitumor drugs; this strategy allows existing and approved clinical drugs to be innovatively repurposed to treat tumors. Based on the similarities between parasitic diseases and cancer, recent studies aimed to investigate the efficacy of existing antiparasitic drugs in cancer. In this review, we selected two antihelminthic drugs (macrolides and benzimidazoles) and two antiprotozoal drugs (artemisinin and its derivatives, and quinolines) and summarized the research progresses made to date on the role of these drugs in cancer. Overall, these drugs regulate tumor growth via multiple targets, pathways, and modes of action. These antiparasitic drugs are good candidates for comprehensive, in-depth analyses of tumor occurrence and development. In-depth studies may improve the current tumor diagnoses and treatment regimens. However, for clinical application, current investigations are still insufficient, warranting more comprehensive analyses.
There exists a close connection between parasitic infections and cancer (1–3). Helminth infections are widespread the world over, and the causative parasites are thought to be responsible for causing cancer in humans (4). Thus far, Schistosoma haematobium, Clonorchis sinensis, and Opisthorchis viverrini have been recognized as clear biological carcinogens (1). The specific carcinogenic mechanism is not yet clear, and the metabolites of catechol estrogens and parasite-derived oxysterols may play an important role (5). Unlike worms, protozoa have not been identified as biological carcinogens; however, certain characteristics of protozoa are similar to those of cancer. In a manner similar to the immune evasion strategies employed by cancers, Trypanosoma cruzi and Leishmania parasites leverage the immune mechanisms to persist in the body and establish a chronic infection (6). Although malaria is the most widespread parasitic disease in the world, it does not seem to be carcinogenic (2). However, the incidence of malaria is positively correlated with mortality in most cancers, with the exception of colorectal, lung, gastric, and several other types of cancer whose mortalities exhibit an inverse correlation with malaria (7, 8). Thus, the relationship between malaria and cancer is worth exploring.
Cancer is the second leading cause of death worldwide and is a major burden of disease (9, 10). However, with proper treatment, many cancers can be cured. Drugs are essential in the treatment of tumors but are often not as effective as required because of drug resistance and low specificity (11). Despite the emergence of highly specific monoclonal antibody drugs, the drugs are unsuitable for a wide range of clinical applications because they have strict requirements for the target (12). Therefore, the development of new antitumor drugs is still urgently needed. However, owing to the similarities between parasitic diseases and cancer as well as the successful clinical administration of antiparasitic drugs for years, it seems feasible to repurpose existing antiparasitic drugs into antitumor drugs. In fact, some antiparasitic and antitumor drugs share the same target, a variety of drugs targeting CDKs, TGR enzyme, tubulin/microtubule system have been confirmed to have dual effects on anti-parasites and anti-cancer (13).
Research on the repurposing antiparasitic drugs for tumors has gradually gained popularity. However, many studies have reported contradictory results. We selected two well-researched antihelminthic drugs and two antiprotozoal drugs and summarized the corresponding research progress to provide direction for further exploration into the repurposing of antiparasitic drugs as antitumor drugs.
Efficacy of macrolide antiparasitic drugs in cancer. Apoptosis is the chief mechanism used by macrolide drugs to kill cancer cells. Macrolides trigger apoptosis through the (1) mitochondrial pathway, (2) cell cycle arrest, and (3) inhibition of the current of Ca2+ ion-activated Cl- channels. Other than apoptosis, macrolides can cause autophagic death of cancer cells by (4) degrading PAK1. When used for cancer cells, macrolides show selectivity for CSCs by (5) inhibiting stem cell genes and inactivating PAK1. Macrolides also reverse the abnormal epigenetics of tumor cells through (6) the combination of PAH2 and SID domain. By binding to the extracellular segment of EGFR, macrolides can (7) inhibit the transcription of P-gp, thereby reversing tumor resistance in which MDR and MASTA1 are also involved.
Avermectins
Commonly used avermectins include avermectin (AVM), ivermectin (IVM), doramectin (DRM), eprinomectin (EPM), and selamectin (SLM). Although all these avermectins show anticancer activity, studies on IVM are more comprehensive than those on other drugs. IVM can regulate the natural progression of tumors via multiple pathways.
Apoptosis is an important mechanism used by IVMs to kill cancer cells. Eukaryotic translation initiation factor 4A isoform 3 (EIF4A3) is an RNA-binding protein involved in the splicing modulation of BCL2L1/Bcl-X and is considered to be closely associated with apoptosis. SILAC-based quantitative proteomic analysis revealed that IVM inhibited the expression of EIF4A3 and 116 EIF4A3-binding mRNAs (15). In LA795 cells, IVM analogs (IVM, EPM, and SLM) can significantly inhibit currents mediated by the transmembrane member 16A (TMEM16A), an endogenous Ca2+-activated Cl- channel closely related to tumorigenesis, thereby inducing apoptosis (16). In addition, IVMs utilize the well-studied pathway of mitochondrial apoptosis to exert their anticancer activity. When acting on HeLa cells, IVM can increase the ratio of Bax/Bcl-2 and induce release of mitochondrial cytochrome c into the cytoplasm, thus stimulating caspase-9/-3-mediated apoptosis (17). In chronic myeloid leukemia and renal cell carcinoma cells, IVM can induce apoptosis by inducing mitochondrial dysfunction (18).
Cell cycle arrest can easily lead to apoptosis. Several studies have revealed that in cancer cells, IVM arrests the cell cycle in different phases by regulating the expression of proteins that control the cell cycle, thereby inducing apoptosis (19–22). Especially in epithelial ovarian cancer, IVM induces apoptosis through multiphasic cell cycle arrest, and exhibits KPNB1-dependent antitumor effects (22). The cell cycle is also the target of many chemotherapeutic drugs, and combinatorial treatment with IVM and clinical drugs is worth investigating. In fact, in multiple in vivo and in vitro experiments, IVM significantly enhanced the efficacy of various drugs, including cisplatin and tamoxifen, but this result still needs clinical verification (20, 23).
Autophagy regulation is another important mechanism underlying IVM action. A study on breast cancer revealed that when IVM was applied to cancer cells, no obvious apoptosis was observed before 24 h of treatment, but the inhibition of growth of these cancer cells during the 24 h was evident. Later in that study, autophagy flux increased during the first 24 h of IVM treatment, and the anticancer effect during this period was reversed when IVM was used to treat cancer cells Beclin 1 or Atg5 knockdown (24). Current studies argue that IVM degrades PAK1 in cancer cells through the ubiquitination pathway, thereby inactivating the AKT-mTOR pathway, which is the key negative regulatory pathway of autophagy (24, 25). Although mechanisms used by IVM require more detailed exploration, it is encouraging that more studies indicate that induction of autophagy may be used as a method of synergistic treatment in clinical tumor chemotherapy, highlighting the potential of IVM as a clinical adjuvant drug (26, 27).
In addition to the anticancer mechanism, the selective functional characteristics of IVMs are also noteworthy. IVMs exhibit more pronounced toxicity toward cancer cells than toward non-cancer cells, which may be related to higher mitochondrial biogenesis in cancer cells (28). More importantly, when acting on cancer cells, IVM still exhibits different levels of cytotoxicity in different cancer cell subgroups. The CD44+/CD24- subpopulation of breast cancer cells have been previously reported to possess stem/progenitor cell properties (29, 30). IVM preferentially inhibits the viability of CD44 +/CD24- subpopulation cells (cancer stem cells (CSCs)) and reduces the expression of stemness genes (NANOG, POU5F1, and SOX2) (31). Current research points out that this may be related to the ability of IVMs to inactivate p21-activated kinase (PAK1), thereby reducing the levels of pStat3 and extracellular IL-6 and inhibiting the formation of CSCs (32). Research on the specific effect of IVM on CSCs is still limited, and many other knowledge gaps exist that require further research. In general, the selective nature of IVMs shows that it is almost non-toxic to non-cancer cells but can effectively inhibit the growth of cancer cells, demonstrating its unique potential as an anticancer drug.
IVMs have also shown anticancer capabilities in many fields other than these mentioned above. Although research on these aspects is rare, it has broadened the scope of exploration of IVMs. The latest research showed that IVMs could reverse tumor resistance. IVM at a low dose that does not produce evident cytotoxicity can bind to the extracellular domain of EGFR, which inhibits the activation of EGFR and its downstream signaling cascade ERK/Akt/NF-κB, thus inhibiting the transcription factor NF-κB and leading to reduction in P-glycoprotein (P-gp) transcription (33). Moreover, in triple-negative breast cancer (TNBC), the targeted disruption of the Sin3 (a master transcriptional scaffold and corepressor that plays an essential role in the regulation of gene transcription and maintenance of chromatin structure) complex by introducing a Sin3 interaction domain (SID) decoy that interferes with PAH2 binding by sequestering SID-containing partner proteins reverted the silencing of genes involved in cell growth and differentiation (34–36). Interestingly, IVM and SLM can be used as small molecule inhibitors of SID peptides that play a similar role to that of Sin3 disruption, indicating that AVMs can also exert anticancer effects by regulating the abnormal epigenetics of tumors (37). Furthermore, the activation of WNT-TCF signaling is implicated in multiple diseases, but there are no WNT-TCF antagonists in clinical use. However, SLM and IVM can reduce the expression of target proteins in this pathway by mimicking dnTCF, further demonstrating the application potential of these drugs (38).
Progress on the Use of Artemisinin and Its Derivatives for Treating Cancer
Artemisinin (ARS) is a 1,2,-trioxane from the Chinese medicinal plant Sweet Wormwood, and since its antimalarial effect was discovered, research on ARS has been continuously focused on. A variety of ARS and its derivatives (ARTs), including dihydroartemisinin (DHA), artemether (ARM), artesunate (ART), and artemisitene (ATT), have emerged because of the advancements of drug modification and synthesis technology. ARS-based combination therapies are established standard treatments for malaria worldwide (84–87). It is currently considered that the heme-irons released by Plasmodium-attacking red blood cells can cleave the endoperoxide bridge of ARS via a Fe (II) Fenton-type reaction, and that free radical intermediates kill the Plasmodia (88, 89). Other pathways are also involved in ARTs antimalarial response (90, 91), but comprehensive research on antimalarial mechanisms remains necessary. Interestingly, as with other natural products, antimalarial properties are not the only benefits of ARTs, and ARTs have shown application value in many diseases, including cancer (Figure 3).
Efficacy of ARTs in cancer. With the help of endogenous peroxides, ARTs can cause oxidative stress by (1) increasing the concentration of ROS, (2) DNA damage, and (3) cell cycle arrest to cause cancer cell death. In addition to the common types of cell death, ARTs increase the concentration of unstable iron ions in cancer cells by (4) regulating a variety of iron-related proteins and IRP1/IRP2, thereby triggering ferroptosis. ARTs strengthen the cancer-killing effect of NK cells by (5) enhancing their degranulation ability and increasing the connection between NK cells and cancer cells. ARTs (6) reduce negative regulation factors (Treg cells and MDSCs) and increase IL-4 and IFN-γ in TME to stimulate T cell immune response.
Progress on the Use of Quinoline Antiparasitic Drugs for Treating Cancer
Similar to ARTs, the design and synthesis of quinoline drugs, which are characterized by a quinoline ring, has been researched for application as antimalarial drugs. Quinolines exert their antimalarial effects during the blood or liver stages of the life cycle of the parasite (140), but different drugs have different mechanisms (141, 142). With the progress in research, an increasing number of quinoline drugs have shown therapeutic effects in other diseases, including cancer (Figure 4).
Efficacy of quinoline antiparasitic drugs in cancer. Lysosomes are an important target of quinoline drugs, and quinolines increase the pH of the lysosome, thereby triggering a variety of cascade reactions. Quinolines can (1) inhibit the degradation of autophagy proteins to block autophagy and (2) stimulate antitumor immune responses by increasing tumor antigens. Mitochondria are also a target for quinolines. Quinolines increase the oxygen concentration in cancer cells by (3) inhibiting the related processes of mitochondrial complex III and cause apoptosis by (4) inducing oxidative stress with the change in mitochondrial membrane potential, (5) inhibiting the phosphorylation level of STAT3 in mitochondria, and (6) introducing double-strand breaks in DNA in cancer cells. (7) Quinolines inhibit the drug delivery mediated by P-gp and BRCP.
Chloroquine
Chloroquine (CQ), a 4-aminoquinoline, has been used as an antimalarial drug for many years and is often recommended to be co-administered with primaquine to prevent recurrence of Plasmodium vivax (143). CQ is currently considered a protonated, weakly basic drug that increases the pH and accumulates in the food vacuole of parasites, thereby interfering with the degradation of host red blood cell hemoglobin, and preventing the growth of malaria parasite (144). The exact mechanism requires further investigation. Similar to other antiparasitic drugs, CQ has also shown potential in the treatment of cancers and other diseases (145, 146).
In summary, antiparasitic drugs are involved in almost all aspects of tumors, including cell cycle, apoptosis, autophagy, ferroptosis, stress, energy homeostasis, immunity, and drug resistance. Besides, when used in combination with existing clinical tumor drugs, many antiparasitic drugs show significant synergistic effects (187–189). However, according to current research results, antiparasitic drugs are still far from being repurposed into antitumor drugs that can be widely used in clinical practice. The current research on the anticancer mechanisms of antiparasitic drugs is still not comprehensive enough, and more thorough research is needed. In addition, antiparasitic and tumor therapy have two different application environments, and many problems remain to be solved. The first problem is drug delivery; the external microenvironment of the parasite-infected foci is relatively normal, whereas the growth of tumors mainly depends on glycolysis, which leads to an acidic external microenvironment. Whether this microenvironment affects the delivery of drugs needs further exploration. The second problem is drug concentration. Different drug concentrations cause different dominant effects. The concentration that has the best anticancer effect and does not cause side effects in the human body needs to be established. The third problem is that although antiparasitic drugs have many advantages, we cannot rule out they might promote tumor growth. Studies have shown that CQ-induced stress in cancer cells can activate NF-κB, thereby conferring transcriptional and phenotypic plasticity to cells, resulting in the reprogramming of cells and allows tumor cells to escape cell death induced by either drug therapy or the immune system (190, 191). Fortunately, for the first two issues, many studies have conducted in-depth study. Increasing evidence shows that nanotechnology-based drug delivery methods yield better therapeutic effects at lower concentrations and might be clinically implemented in the near future (192–198). Successive preclinical studies and clinical trials have clarified in detail the therapeutic effects of different drug concentrations and various possible side effects. However, there are very few studies have investigated the cancer-promoting effects of antiparasitic drugs. However, this may be very important for us to fully understand the role of antiparasitic drugs in tumors. After understanding these cancer-promoting effects, with the help of modern drug modification and improvement technologies (199–202), it can greatly accelerate the real application of antiparasitic drugs in clinical cancer treatment. More problems are likely to be encountered as research and practice progress. Nevertheless, it is undeniable that antiparasitic drugs indeed have great potential for development as broad-spectrum, clinically applicable antitumor drugs.
Li YQ, Zheng Z, Liu QX, Lu X, Zhou D, Zhang J, Zheng H, Dai JG. Repositioning of Antiparasitic Drugs for Tumor Treatment. Front Oncol. 2021 Apr 29;11:670804. doi: 10.3389/fonc.2021.670804. PMID: 33996598; PMCID: PMC8117216.
DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.












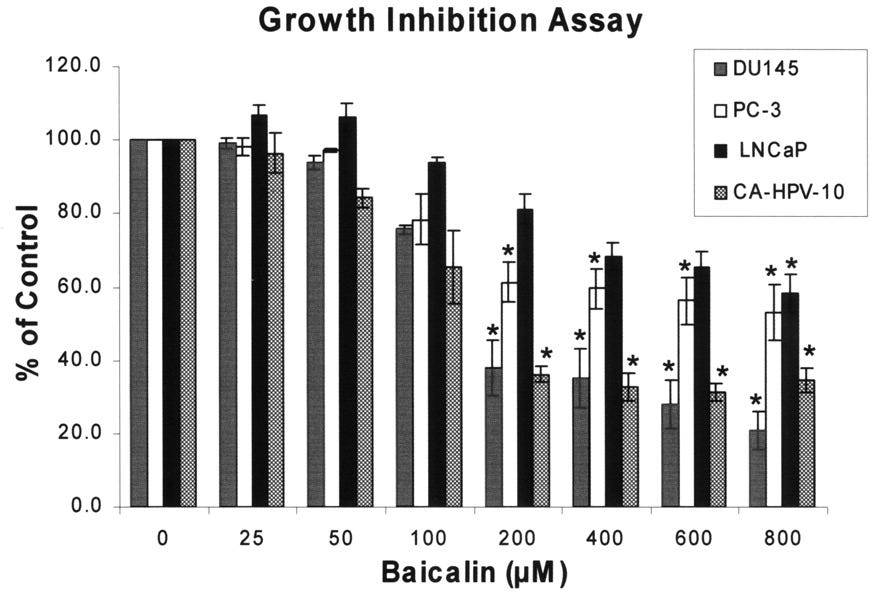
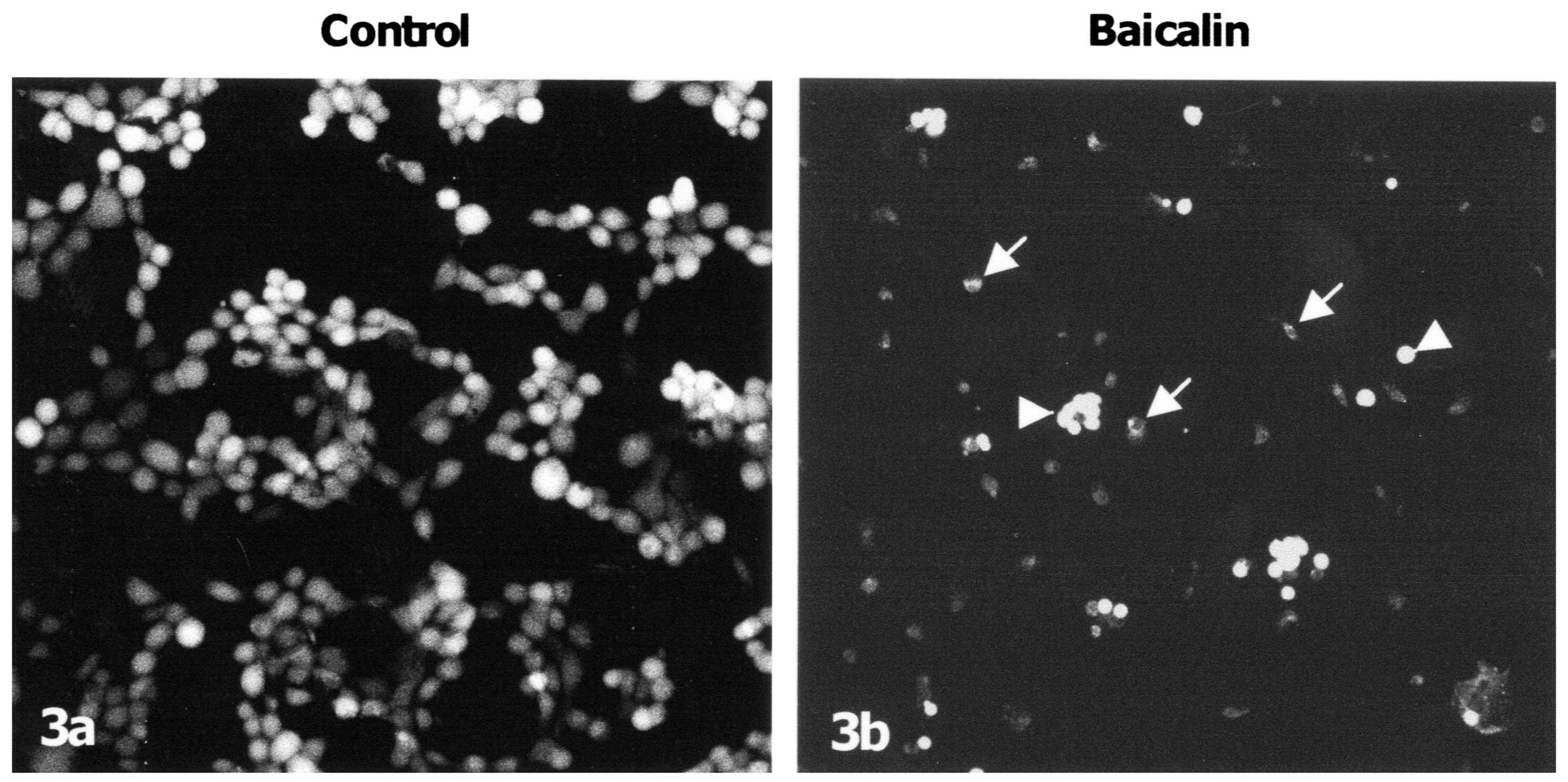
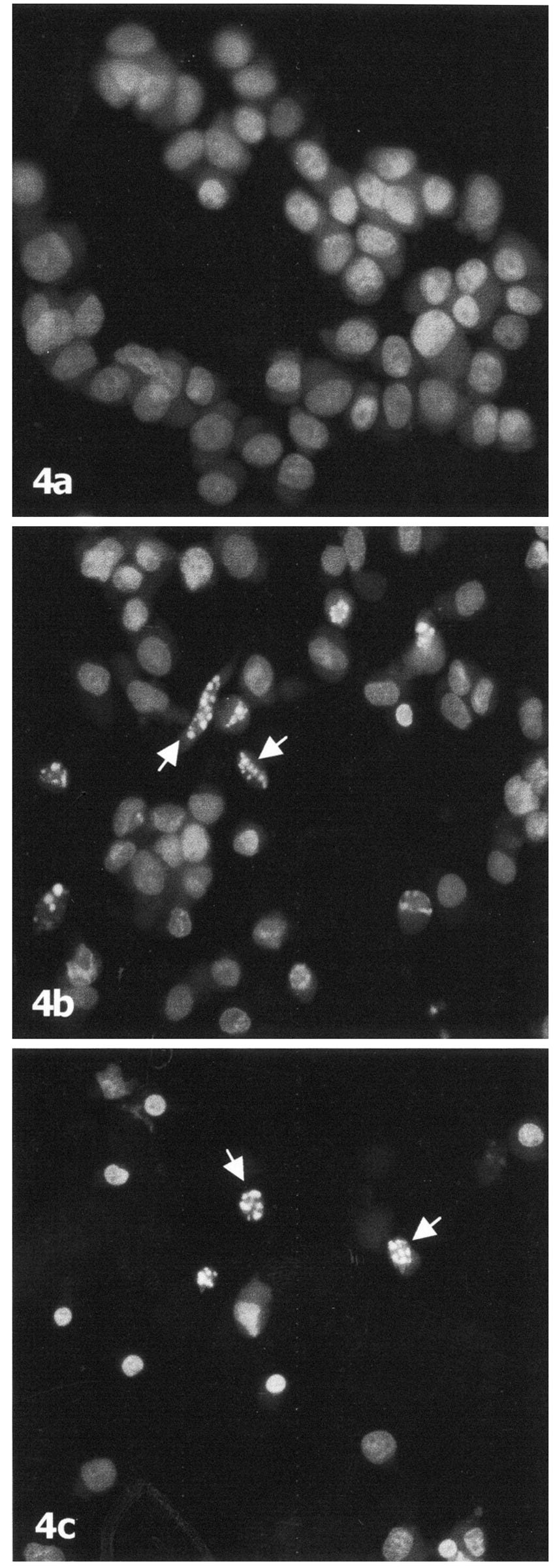






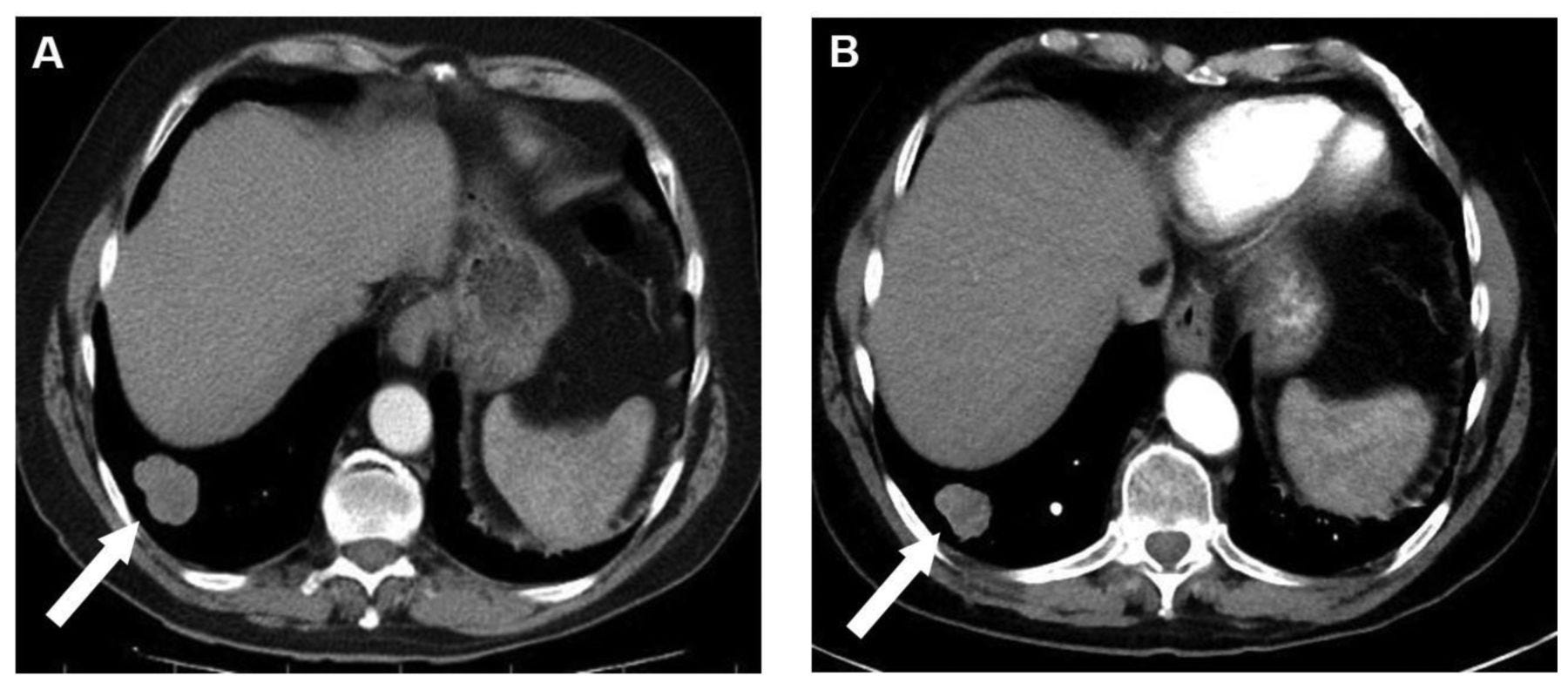





















{kind=link}
{kind=link}




Gosh, incredible library to save and reference - a mere thank you doesn’t cover it!
What an extraordinary post! What a gift. Thank you. There is do much to digest.