
IgG4 class switching and Immunoglobulin G4-related aortitis - Part IV: A deep dive into mechanisms
Reading time:
short story - novelette - novella - novel - PhD thesis - War and Peace - U.S. Tax Code
Also available with the translator, 🇫🇷 🇪🇸 🇩🇪 🇯🇵 etc:
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.

Contents

Introduction
In Part I I reviewed 3 case reports of IgG4-RA and myocarditis; Part II reviewed 7 case reports of other diseases associated with IgG4 class switching after COVID-19 vaccination; and Part III started to review the mechanisms behind IgG4-RA, with a deep dive into macrophage priming via the fragment chain receptors(FcRs)
In Part IV we will see if the latest vaccines have solved the problem, investigate mechanisms of inhibition, macrophage polarisation, Fc receptor interactions, and a microRNA that keeps turning up in cancers and autoimmune disorders as reliably as finding a politician on the payroll of big pharma.
Discussion
An ongoing problem
The MHRA rubber-stamped an Omicron XBB.1.5 specific monovalent engineered mRNA shot at the end of 2023. Unfortunately, a study the following year by Jain et al. (2024) showed that it was just as potent at inducing class switch recombination (CSR) to IgG4 as the previous mRNA vaccines.1
The lie being presented (emphasis mine throughout):
An adapted COVID-19 vaccine from Moderna (Spikevax) that targets the Omicron XBB 1.5 subvariant, has today been authorised by the Medicines and Healthcare products Regulatory Agency (MHRA) after it was found to meet the UK regulator’s standards of safety, quality and effectiveness.
The vaccine has been approved for use in individuals from 6 months of age.
The adapted vaccine works in the same way as the original vaccine by causing the immune system (the body’s natural defences) to produce antibodies and blood cells that work against the virus, so giving protection against COVID-19. All approved adapted COVID-19 vaccines help to improve the protection obtained from earlier doses of the vaccine and help give longer-term protection against getting seriously ill from COVID-19.
From: “MHRA approves Moderna’s adapted COVID-19 vaccine (Spikevax) that targets Omicron XBB.1.5” (15th Sept. 2023)
The reality is that even the new variant monovalent vaccines still induce class-switch recombination (CSR), with high titres of IgG4.
Key takes from “XBB.1.5 monovalent booster improves antibody binding and neutralization against emerging SARS-CoV-2 Omicron variants“2 by Jain et al. (5th Feb. 2024):
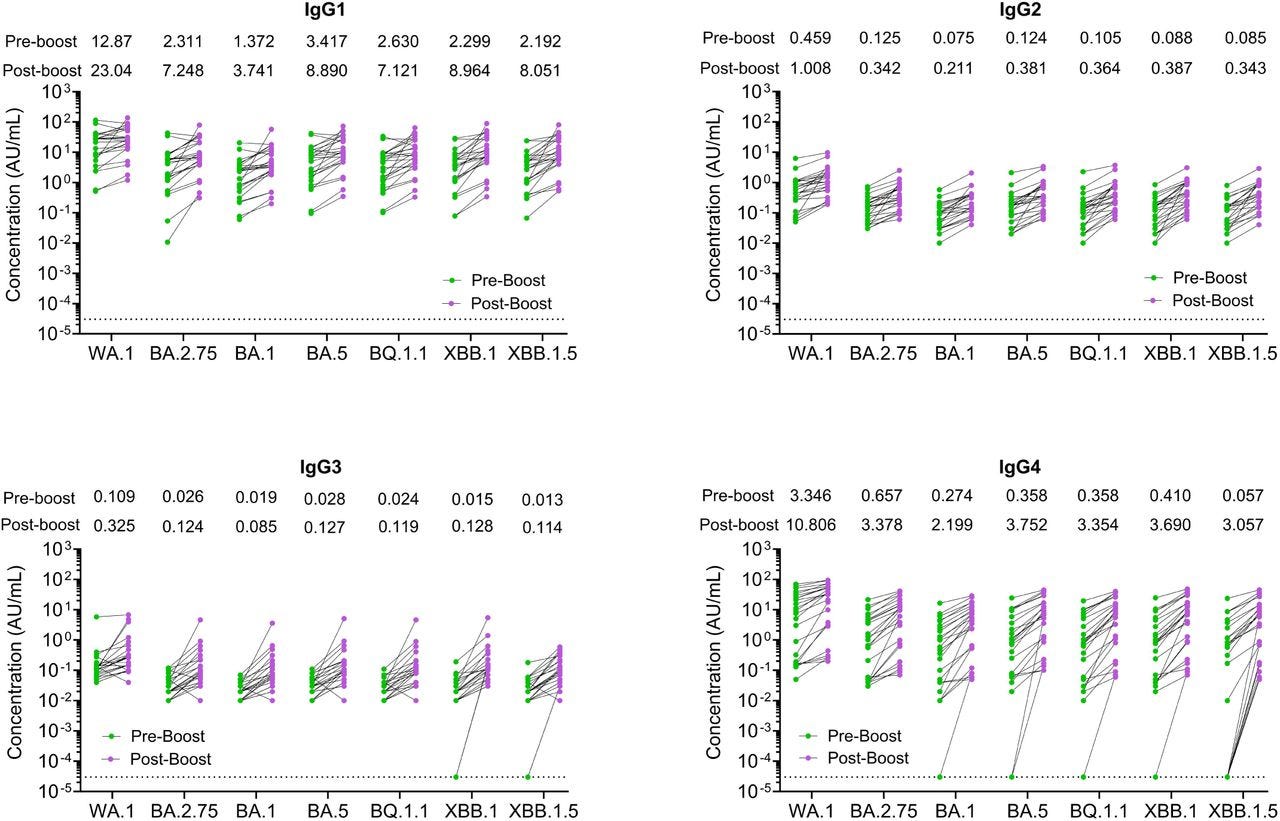
… Following the XBB.1.5 monovalent booster, IgG1, IgG2, IgG3, and IgG4 binding to the WA1 spike protein increased by 1.8-fold, 2.4-fold, 3-fold, 3.2-fold, respectively (Fig. 3).

From “Figure 3. SARS-CoV-2 IgG subclass binding to SARS-CoV-2 variants following the XBB.1.5 moovalent booster dose. IgG antibody subclasses responses against SARS-CoV-2 spike proteins were measured by an electro-chemiluminescent multiplex immunoassay using the MSD platform and reported as arbitrary units per ml (AU/mL) as normalized by a standard curve using a monoclonal antibody. Shown are the spike-binding IgG subclasses antibody titers against the WA1 and the Omicron subvariants for the 24 participants before (green symbols) and after (pink symbols) receiving XBB.1.5 booster immunization. The dotted line in the spike binding assays represents the limit of detection.” Source: https://www.biorxiv.org/content/10.1101/2024.02.03.578771v1.full … Further research is therefore needed to evaluate the mechanisms of the preferential IgG4 class switching induced by the XBB.1.5 monovalent mRNA vaccine, which is relevant for designing future seasonal SARS-CoV-2 booster immunizations.
It has been speculated that the emergence of isotype-switched memory B cells is consistent with a more robust and durable germinal center (GC) response induced by mRNA-based vaccines25.
These findings contrast with the predominant induction of IgG1 and IgG3 after natural SARS-CoV-2 infections26. IgG4 has limited functional activity due to structural features that impair its binding to C1q and Fc receptors that mediate opsonization and antibody-depending cellular cytotoxicity27.
These features likely make IgG4 a blocking or suppressing antibody28. Further investigations are therefore required to define the role of anti-spike IgG4 induced by XBB.1.5 monovalent booster vaccine in protection or its impact on the response to future booster immunizations.
What about bivalent shots?
If you were hoping that the newer engineered mRNA products could reverse IgG4 CSR then unfortunately it’s not good news.

Hat tip to Dr Alberto Rubio-Casillas for sharing a very recent study investigating this exact issue.
In brief, their research seems to confirm that the elderly are at increased, not decreased risk of CSR, as perhaps the authors originally thought.
Key takes from “Repeated COVID-19 mRNA vaccination results in IgG4 class switching and decreased NK cell activation by S1-specific antibodies in older adults”3 by Gelderloos et al. (14th Sept. 2024):
Results
Spike S1-specific IgG subclass concentrations (expressed in arbitrary units per mL), antibody-dependent NK cell activation, complement deposition and monocyte phagocytosis were quantified in serum from older adults (n = 38–50, 65–83 years) at one month post-second, -third and -fifth vaccination.
Subclass distribution in serum was compared to that in younger adults (n = 64, 18–47 years) at one month post-second and -third vaccination.
It actually gets worse the older you are, despite the onset of immunosenescence and a lifetime’s exposure to other antigens, such as cross-reactive coronaviruses:
Compared to younger individuals, older adults showed increased levels of IgG2 and IgG4 at one month post-third vaccination (possibly related to factors other than age) and a further increase following a fifth dose.
It’s not just a paper exercise either. The more IgG4 abs you have relative to IgG1 the less your natural killer cells get activated by SARS-CoV-2 and the more impaired your complement system becomes:
The capacity of specific serum antibodies to mediate NK cell activation and complement deposition relative to S1-specific total IgG concentrations decreased upon repeated vaccination. This decrease associated with an increased IgG4/IgG1 ratio.
Conclusions
In conclusion, these findings show that, like younger individuals, older adults produce antibodies with reduced functional capacity upon repeated COVID-19 mRNA vaccination.
The reason for our reviews and these Substacks, along with our recommendations for more research and clinical assessments:
Additional research is needed to better understand the mechanisms underlying these responses and their potential implications for vaccine effectiveness. Such knowledge is vital for the future design of optimal vaccination strategies in the ageing population.
… Next, we assessed the SARS-CoV-2 spike S1-specific concentrations of IgG1, IgG2, IgG3 and IgG4 using a bead-based multiplex immunoassay (Fig. 2).
This has all the makings of a public health nightmare, especially if, as the data appears to suggest, immune response is not returning to normal with time:
Whereas only a (small) proportion of participants has detectable levels of IgG2 and IgG4 post-second vaccination (58% and 2.1%, respectively), virtually all participants have detectable levels of these subclasses after the third vaccination (100% and 94%, respectively).
Again, if you were infected prior to vaccination, this appears to be protective. My only concern is that, anecdotally, many of us were infected in late 2019/20 prior to vaccination. Did you need to be infected close to the time of vaccination for it to prevent CSR?
Despite the very limited group size (n = 4), we observed that those older individuals that were excluded from the overall analysis because they had been infected prior to receiving their first COVID-19 vaccination displayed remarkably reduced levels of IgG4 and IgG2 following repeated vaccination (Fig. 3 and Supplementary Figure S2).
We did not observe notable differences in IgG subclass concentrations between female and male participants (Supplementary Figure S3).

Since we observed a striking induction of S1-specific IgG4 antibodies in older adults we wondered whether this could be associated with differences in the functional capacity of the antibodies relative to total S1-specific IgG concentration.
Yes! More IgG4 Abs = less functional anti-Spike Abs:
Our data suggest that, in line with the known functional properties of IgG4, higher S1-specific IgG4/IgG1 ratios – i.e. relatively higher concentrations of IgG4 – indeed associate with a lower relative capacity to activate NK cells and complement deposition (Fig. 6A).
From these graphs it is evident that the darker datapoints (lower IgG4/IgG1 ratios) tend to localize above the line representing the average correlation between functionality and concentration, while the lighter datapoints (higher IgG4/IgG1 ratios) tend to lie below this line, especially for ADNKA.
The S1-specific phagocytosis capacity relative to total IgG concentration did not clearly associate with the IgG4/IgG1 ratio.
ADNKA: antibody-dependent NK cell activation (Left-hand column).
Interpretation: A higher ratio of IgG4 to IgG1, in yellow, is associated with markedly reduced natural killer cell activation:

Previously, we have also observed a decline in antibody-dependent NK cell activation relative to antibody concentration over time following primary SARS-CoV-2 infection in children and adults [33].
Although we did not evaluate subclass levels in that study, the decline in that setting was likely due to waning of virus-specific IgG3 antibodies, which are known to be superior mediators of ADNKA but have a short half-life compared to other subclasses [19].
You cannot blame the virus for this:
In addition, it is highly unlikely that significant amounts of IgG4 have been produced in response to a primary viral infection.
In contrast, in the current study the decline in ADNKA relative to antibody concentration does not appear to relate to the presence of IgG3, as the median levels for this subclass actually increase from the post-third to post-fifth vaccination timepoint. The potential role of IgG4 in the decline in ADNKA relative to antibody concentration is in line with the known functional characteristics of IgG4 [18, 19].
I would rebut this conclusion, as the vaccines never were effective at preventing infection or transmission in the first place, and breakthrough infections are a common occurrence:
At present, it remains unclear to what extent (if any) the occurrence of virus-specific IgG4 will affect vaccine effectiveness, which thus far appears to remain sufficient [41, 42].
Their own study findings also confirm that this is nonsense:
As expected based on earlier work, our study confirms that increased levels of IgG4 associate with reduced Fc-mediated effector functionality [6, 19]. Considering that in addition to virus neutralization (which is not affected by IgG4 induction), there is increasing evidence suggesting that these Fc-mediated effector functions contribute to immunological protection from disease [20,21,22,23,24,25,26,27,28, 43], one might expect that IgG4 induction is not beneficial for vaccine effectiveness.
They are clutching at straws here. Impaired immune responses to a chimeric virus with HIV inserts are the last thing you want. I suspect it would do nothing to prevent cytokine storm either, as this affects individuals with high viral loads, possibly due to overactive regulatory COVID-19 regulatory T cells (Tregs), which can impair antiviral responses4:
Alternatively, IgG4 might play a beneficial role in reducing the inflammatory potential of continuously increasing IgG levels upon repeated vaccination [18].
I cannot disagree with this:
Either way, it will be imperative to follow this development in larger population studies in which breakthrough infections and symptoms are duly recorded, especially in light of potential additional booster vaccinations.
In conclusion, we have shown that older adults, like younger individuals, are inclined to develop IgG4 responses upon repeated COVID-19 mRNA vaccination and that increased IgG4 levels associate with a relative reduction in Fc-mediated effector functionality.

Mechanisms of inhibition
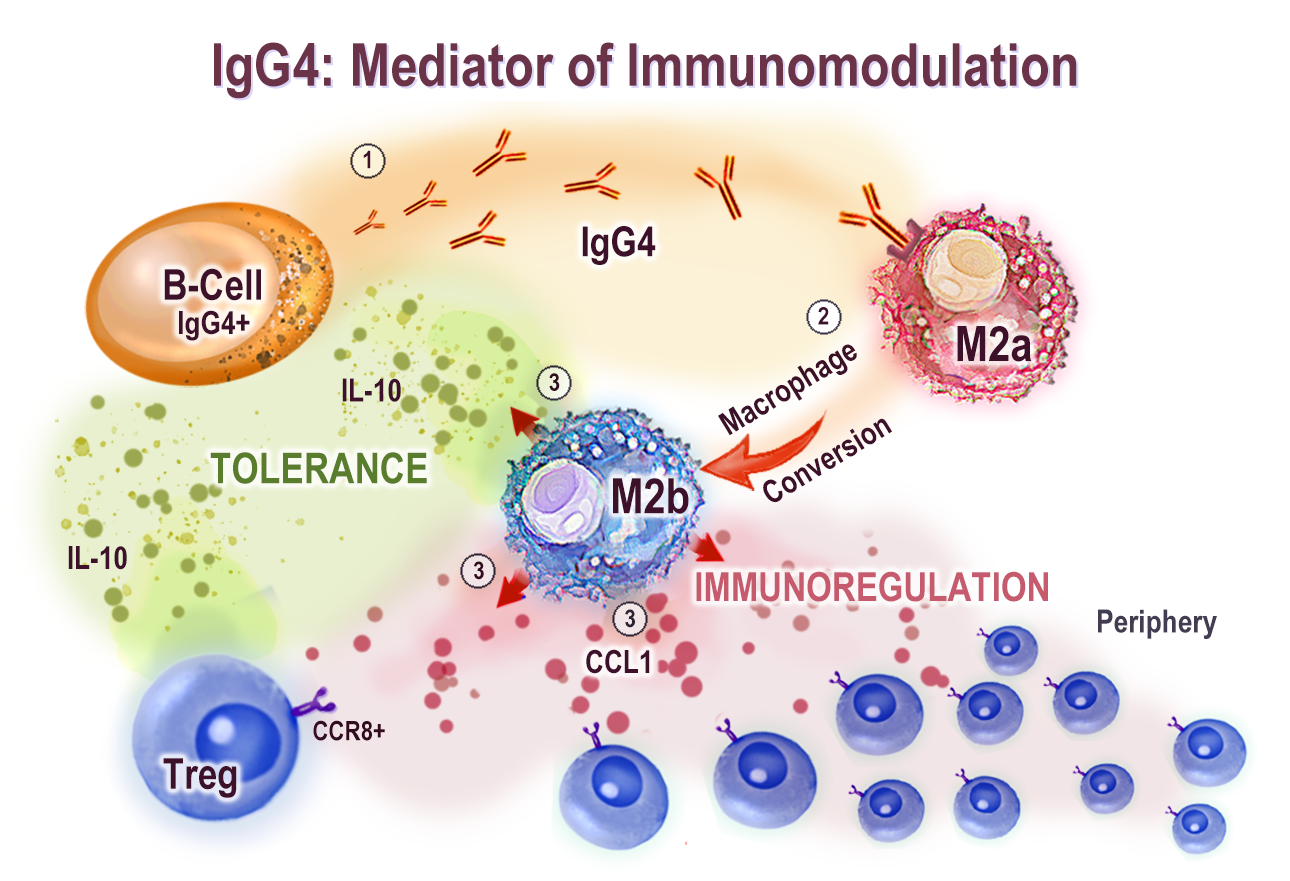
A great introduction to the immunomodulatory properties of IgG4 comes from “The Role of IgG4 in the Fine Tuning of Tolerance in IgE-Mediated Allergy and Cancer” (2020) by Bianchini et al.5:
Abstract
Among the four immunoglobulin G (IgG) subclasses, IgG4 is the least represented in serum of a healthy human and it is considered an “odd” antibody. The IgG4 antibody has unique structural features that affect its biological function. These include the ability to undergo antigen-binding fragment (Fab)-arm exchange, to create fragment crystallizable (Fc) – Fc binding with other IgG4 and other IgG subclass antibodies, have a unique affinity profile for Fc gamma receptors (FcγRs) and no binding to complement component C1q.
Altogether, these characteristics support anti-inflammatory roles of IgG4 leading to immune tolerance. Under conditions of chronic antigenic stimulation and Th2-type inflammation, both tissue and serum IgG4 levels are increased.
This review seeks to highlight how in allergen immunotherapy IgG4 can confer a protective role as a “blocking” antibody and safeguard from subsequent allergen exposure, while IgG4 can confer immunomodulatory functions to support malignancy.
While Th2 conditions drive polarization of macrophages to the M2a subtype, chronic antigen stimulation drives B cell class switching to IgG4 to further support phenotypical macrophage changes towards an M2b-like state.
M2b-like macrophages can secrete chemokine (C-C motif) ligand 1 (CCL1) and interleukin-10 (IL-10) to support regulatory cell recruitment and to further shape a tolerogenic microenvironment. Thereby, IgG4 have a Janus-faced role, favorable in allergy but detrimental in cancer.
Macrophage polarisation to M2b
From “M2b macrophage polarization and its roles in diseases”6 (2018) by Wang et al.:
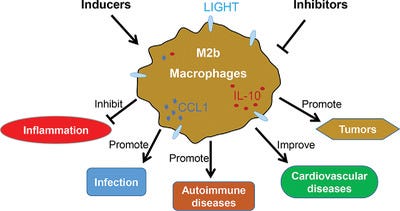
Macrophages commonly exist in two distinct subsets: classically activated macrophages (M1) and alternatively activated macrophages (M2). M2b, a subtype of M2 macrophages, has attracted increasing attention over the past decade due to its strong immune-regulated and anti-inflammatory effects.
A wide variety of stimuli and multiple factors modulate M2b macrophage polarization in vitro and in vivo. M2b macrophages possess both protective and pathogenic roles in various diseases. Understanding the mechanisms of M2b macrophage activation and the modulation of their polarization might provide a great perspective for the design of novel therapeutic strategies.
M2 macrophages can be polarized by several stimulatory factors, which include cytokines (IL-4, IL-10, and IL-13), glucocorticoids, immune complexes (IC) and LPS. Different subtypes of M2 macrophages can be induced by different stimulatory factors.
You need to be very careful when using therapeutics that change macrophage polarisation:
As an M2 subtype, M2b macrophages, also known as regulatory macrophages, can be induced upon combined exposure to IC and TLR agonists or by IL-1R agonists and express high levels of CCL1 and TNF superfamily member 14 (TNFSF14).2, 4, 7, 14, 23, 24
They can be proinflammatory too:
In addition to proinflammatory cytokines (IL-1β, IL-6, and TNF-α), M2b cells also express and secrete substantial amounts of the anti-inflammatory cytokine IL-10 and low levels of IL-12, which is the functional converse of M1 cells.2, 4, 24 Based on the expression profile of cytokines, chemokines, and other secreted factors, M2b macrophages regulate the breadth and depth of the immune response and the inflammatory reaction.2
… or they may promote cancer and infectious diseases:
In cancer and infectious diseases, M2b macrophages promote tumor development and parasite, bacterial, and fungal infections by blunting the immune and inflammatory response.25-31
Moreover, M2b macrophages can reduce spinal cord injury (SCI) and myocardial ischemia/reperfusion (IR) injury, and contribute to recovery from these injuries.6, 24, 32
Despite various important roles in many diseases, the definite and specific molecular markers of M2b macrophage have not yet been unified and established to date.
Comparisons can be made between IgG4-RD and radiation sickness:
3.4 Modulation by radiation
Acute radiation exposure can cause lethal injuries to the hematopoietic and gastrointestinal systems.129 After 5–9 Gy whole body radiation, the macrophages of mesenteric lymph nodes (MLNs) polarize toward the M2b phenotype.49, 50
This subtype of macrophages is not converted to M1 macrophages in response to stimulation by E. faecalis antigens and inhibits the conversion of macrophages from resident M0 to M1.50
A further study has shown that miR-222, induced by whole body radiation, reduces the expression of GAS5, resulting in an increase in CCL1 levels and macrophage conversion to M2b.49
Suppressing CCL1 reversed the M2b polarisation:
Moreover, after treatment with CCL1 antisense oligodeoxynucleotides (CCL1-ODN, which can decrease the mRNA level of CCL1), M2b macrophages disappear in the MLNs of radiated mice, and M1 is generated in the MLNs of these mice following E. faecalis stimulation.50 However, it is unclear how radiation induces the expression of miR-222.
In addition, some reports have shown that thoracic radiotherapy increases cardiovascular events and atherosclerosis development in patients with cancer.11, 130, 131
Interestingly, local 14 Gy radiation in ApoE−/− mice results in a larger number of M1 and smaller number of M2 macrophages in atherosclerotic lesions.132 Additionally, radiation reduces the phagocytic capacity of M2 macrophages, likely contributing to the increase in apoptotic cells and to the shift of macrophage polarization toward a proinflammatory M1 phenotype in vitro.132
These observations indicate that during different diseases, different doses of radiation have various effects on the macrophages in different tissues. Due to the extensive application of radiotherapy in cancer patients, it will be important to investigate the effect of radiation on macrophage phenotype to improve the radiotherapy efficacy in further studies.
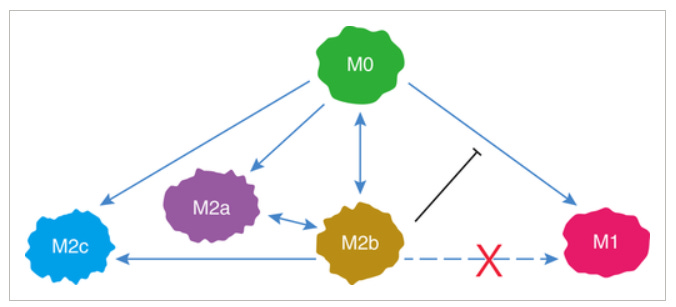
They can contribute to the development of lupus:

M2b macrophages play an important role in inflammatory diseases, especially in autoimmune or autoinflammatory diseases. Studies have shown that M1 macrophages play an important inflammatory role in the pathogenesis of systemic lupus erythematosus (SLE), and M2b macrophages actually have a direct role in causing SLE.
They can, as above, be protective against CVD:
In myocardial disease, our recent study indicated that M2b macrophages (IL-10+, LIGHT+, F4/80+) have cardioprotective effects.24 We induced BMDM polarization to M2b macrophages by exposure to LPS and IC. Then, we transplanted M2b macrophages into the myocardial I/R injury zone and found that the serum cardiac troponin I (cTnI) level, infarct area, and apoptosis index were decreased in the group of transplanted M2b macrophages.
The cardioprotective effect of the transplanted M2b macrophages occurs via their reduction of NF-κB signaling activation and of the up-regulated expression of A20 in heart caused by I/R injury.
TAMs: A percentage of tumour-associated macrophages have been found to be M2b’s:
Accumulating data have shown that M2b monocytes/macrophages can promote the growth, invasion, and recurrence of cancers in vitro and in vivo.25, 28, 162, 163 These observations indicate that M2b macrophages may be one important component of TAMs, demonstrating different quantities in different cancers.
Further studies are needed to investigate the percentage of M2b in TAMs of different tumors and the molecular mechanism by which M2b macrophages promote tumor development.
Fc receptor shenanigans
Another useful reference is a review from 2015 by Li & Kimberley: “Targeting the Fc receptor in autoimmune disease”7.
As per the title, if autoantibodies are presented to the Fc receptors, then autoimmune disorders can result, as the body attacks itself. In this review, the authors discuss these mechanisms and how we can target them with therapeutics. This isn’t the focus of this Substack, but they do provide a great introduction to the different FcRs and which one to target to suppress an autoimmune response. It also signposts us to how IgG4 does the same.
Its very useful to us to prevent auto-immune disorders or allergic responses to bee stings say. But when IgG4 allows us to get re-infected with high viral load SARS-CoV-2, or if its suppressing anti-tumour responses then this could prove fatal or lead to reduced life expectancy.
Key takes:
FcRs are classified by the Ig isotype of their ligands, such as IgG, IgA, IgE and IgM.
IgG is the most abundant class of antibody in circulation, constituting 75% of immunoglobulin in serum.
The FcγR is a group of surface glycoproteins encoded by eight genes located in chromosome 1q21-23 that bind to the Fc portion of IgG. Besides interacting with IgG, FcγRs also function as receptors for innate immune opsonins (CRP and SAP) and provide a link between innate and acquired immunity.
Based on structural homology and differences in affinity this family is further divided into three subfamilies: FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16) (Figure 1A).
Note:
FcγRIIb is the only inhibitory receptor. The FcγRII family of leukocyte FcRs is also known as “cluster of differentiation 32”, or CD32.
FcγR functions either as activating receptors (FcγRI, FcγRIIa/c, FcγRIII) or inhibitory receptors (FcγRIIb), as they signal through immune tyrosine activating or inhibitory motifs (ITAM or ITIM) to elicit or inhibit immune functions.
ITAM = activating motif (in green); ITIM = inhibitory motif (in red):

FcγRI.
With three extracellular Ig-like domains, FcγRI is the only FcγR with high affinity (Ka~109 M−1), enabling it to bind monomeric IgG strongly. FcγRI family has three genes (FCGRIA, FCGRIB and FCGRIC) but only FcγRIa, product of FCGRIA, has been identified as a full length mature receptor [3]. FcγRI has been found on surface of monocytes, dendritic cells (DC), macrophages and neutrophils when they are primed with interferon-γ (IFN-γ) or granulocyte colony stimulating factor (G-CSF) [4].
FcγRII.
FCGR2 and FCGR3 gene variants are located in the classical low affinity Fc receptor cluster on human chromosome 1q23. FcγRII subclass is composed by three genes (FCGR2A, FCGR2B and FCGR2C) which encode FcγRIIa, FcγRIIb and FcγRIIc. Being expressed on monocytes, neutrophils, B cells and NK cells, FcγRII (CD32) is the most widely distributed FcγR with low binding affinity to IgG [5].
IC = immune complex. In other words, IgG4 has a higher affinity for mixed and combined antibodies rather than simple (single) IgG abs:

With two extracellular Ig-like domains, FcγRII has low binding affinity for monomeric IgG, but readily binds IgG aggregates and IC.
While FcγRIIa and FcγRIIc contain an ITAM, FcγRIIb distinguishes itself as the only inhibitory Fcγ receptor by comprising an ITIM on its intracellular domain.
Table 1, shows the distribution of FcγRIIb and its binding preferences:

The review then describes the attributes of the other receptors.
Of particular note are the signalling properties.
A phosphatase is an enzyme that accelerates the transfer of phosphate groups to other compounds. When this happens in the ERK pathway, inactivation occurs, leading to growth inhibition or an apoptotic response in various cell types.
A kinase does the opposite to this (by phosphorylation), and is typically involved with enzyme actvation.
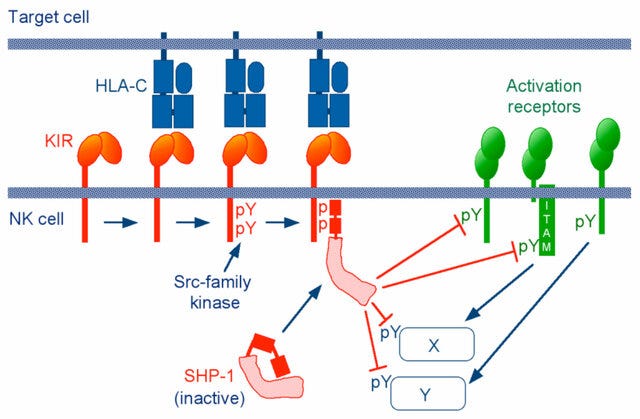
Antibody mediated cell activation threshold is tuned by activating and inhibitory FcRs. Upon crosslinking of the activating receptors, the tyrosines in ITAM are phosphorylated by Src family kinases such as Lyn and Fyn, which then leads to the recruitment of the cytosolic protein kinase Syk [24, 25].
Consequences of the activating signaling include oxidative burst, degranulation, phagocytosis, cytokine production, and antibody-dependent cell-mediated cytotoxicity, or in general, the initiation of inflammation and tissue damage in the development of autoimmunity, as well as immune complex elimination.
Of relevance to IgG4 signalling:
BCR = B-cell receptor.
SHIP = SH2 domain-containing inositol phosphatase.
SHP-1 and SHP-2 = SH2 domain-containing phosphatases 1 and 2.
Mode of action and links to autoimmune disorders and cancer:
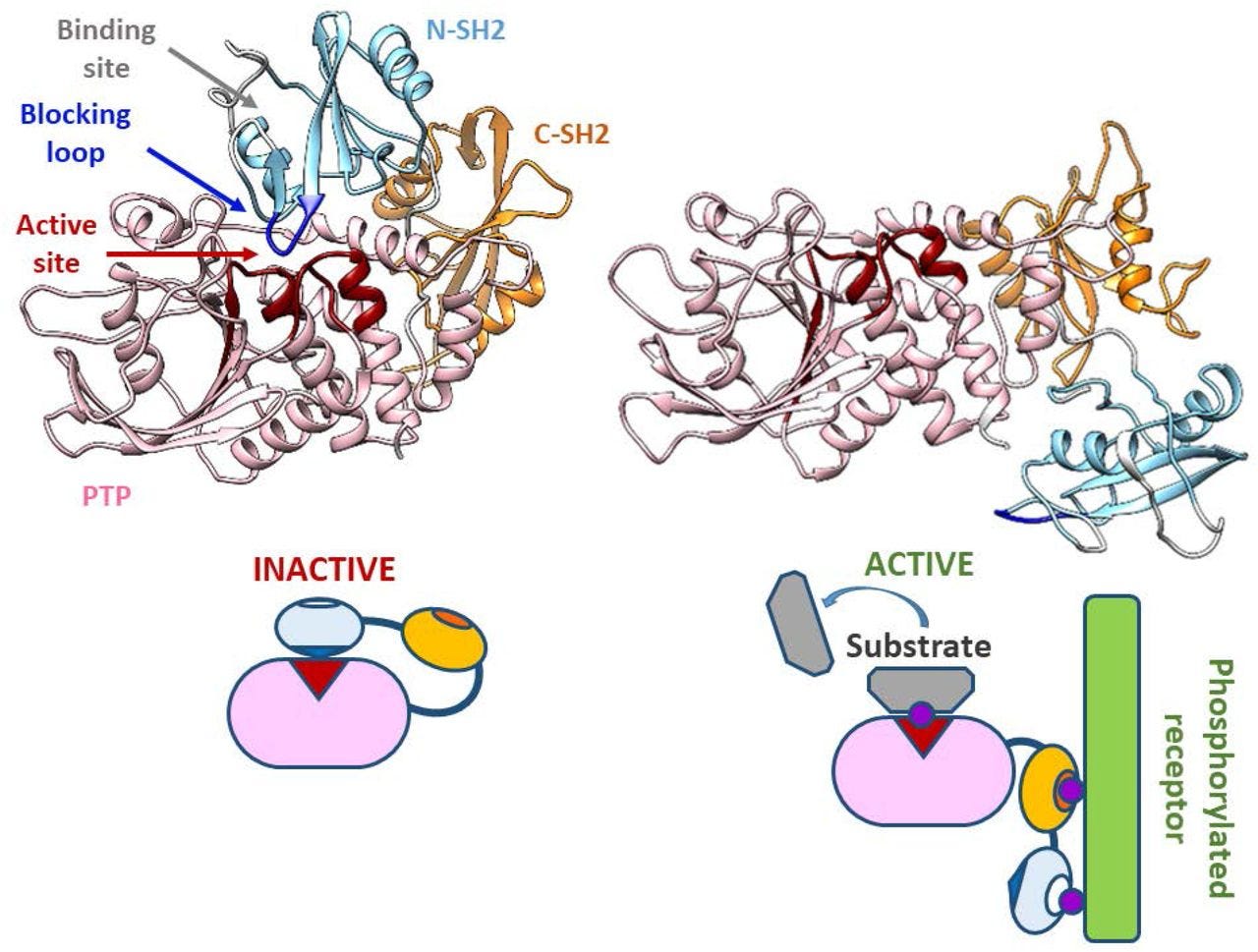
Two tyrosine residues at the C-terminus of SHP-1 (Y536 and Y564) are phosphorylated by various stimuli. The phosphorylation of tyrosine residues modifies the function and activity of SHP-1 depending on the property of the stimulus. SHP-1 exists in an auto-inhibited conformation (Yang et al., 2003). The N-SH2 domain binds tightly to the PTP domain and blocks substrate access to the catalytic domain, thus keeping the enzyme in its inactive conformation. On the contrary, when the phospho-peptide is bound to the C-SH2 domain, the N-terminal SH2 domain is released from the PTP domain, which relieves this autoinhibition and catalytically activates the enzyme (Figure 1B). The “moth-eaten” mice demonstrated a deficiency of SHP-1 and were affected by many hemopoietic disorders, including autoimmune hyperactivation of macrophages, which that suggested a defect in negative regulation (Shultz et al., 1997; Tsui et al., 2006).
The SHP-1 gene has two promoters: the distal promoter, which is only present in epithelial cells, and the proximal promoter, which is active in both epithelial and hemopoietic cells. Therefore, under normal conditions, expression levels of SHP-1 vary between epithelial and hematopoietic cells. This concept is supported by an aberrant level of SHP-1 in cancers (Tsui et al., 2006). In hematopoietic cancers, such as myeloma, promoter methylation inhibited SHP-1 expression. Conversely, SHP-1 has been found to be overexpressed in epithelial cancer, such as breast cancer (Tsui et al., 2006).
From: “Strategy for Leukemia Treatment Targeting SHP-1,2 and SHIP“ (2021)
In other words, phosphorylated ITIM inhibits MAPK signalling by changing the conformation of SHP-1 & SHP-2 to block access to the active sites.
Graphical representations of the above:
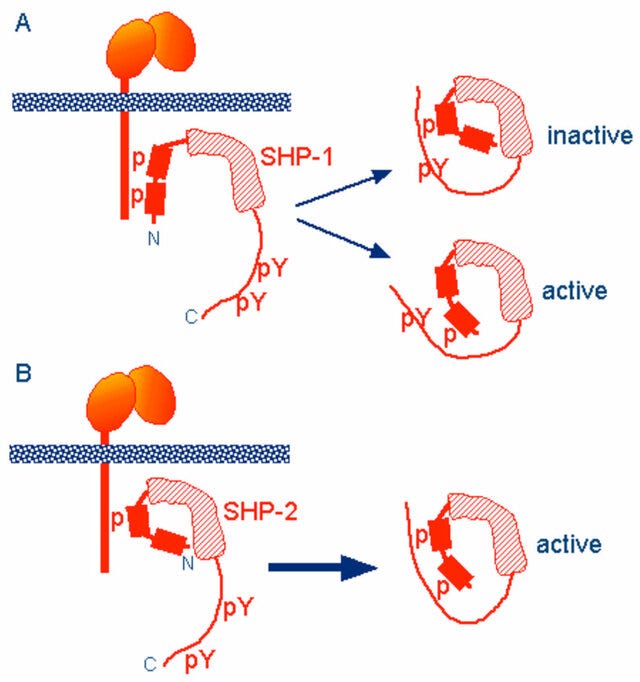
A Richardson (or ribbon) diagram showing the mechanisms of inhibition in greater detail:
When the inhibitory FcγRIIb is co-engaged with activating Fc receptors or the BCR, the tyrosine-phosphorylated ITIM recruits the SH2 domain- containing phosphatases SHIP or SHP-1[26].
Btk is another important kinase in the BCR signalling pathway. IgG4 binding to FcγRIIb also helps to inhibit its effects:
In B cells, the cascade extends to dissociate Btk and PLC-γ from the BCR signalosome and reduce calcium influx [27]. Through its ITIM, FcγRIIb serves as a negative regulator of immune complex-triggered inflammatory cascades.
If FcγRIIb signalling is suppressed, or lessened due to mutations, autoimmune disorders may result.
(SLE = lupus).
The only inhibitory FcR, FcγRIIb, serves as a critical negative regulator in immune complex driven reactions. In mice lacking FcγRIIb, autoimmune symptoms are exacerbated, and a partial restoration of FcγRIIb expression on B cells was able to rescue mice from developing an SLE-like phenotype [46–48].
The important suppressor role of FcγRIIb in the development of autoimmunity is further supported by multiple observations in human that FcγRIIb polymorphisms are associated with SLE by affecting either signaling or expression level [49–52]. FcγRIIb on memory B cells is also reported to be dysregulated in SLE [53, 54].
As a single target then treating it should be easy, but it is challenging to get an antibody to target the ITIM FcγRIIb without accidentally stimulating the ITAMS FcγRIIa or FcγRIIc and making the condition worse:
These unique inhibitory features mark FcγRIIb as an important candidate for therapeutic targets in autoimmunity. However, for many years antibody therapy against FcγRIIb had been dampened by the great difficulty in distinguishing FcγRIIb from FcγRIIa due to their highly homologous extracellular sequences.
Of relevance to mechanisms of immunosuppression by IgG4 :
The level of FcγRIIb expression on monocyte-derived dendritic cells (mDCs) sets the threshold for TLR4-mediated cell activation in response to IC stimulation. FcγRIIb inhibits TLR4-mediated DC maturation, reduces secretion of pro-inflammatory cytokines, which in turn inhibits T cell proliferation, and primes T cells to produce anti-inflammatory Th2 cytokines [152].
The machinery for this effect may be interacting signaling components. Activation of FcγRIIb ITIM recruits phosphorylated SHIP and down-regulates PI3K and Akt, which are key molecules in TLR4 signaling. Consistent with the experimental data, a high level of expression of FcγRIIb on DCs is associated with reduced disease activity and quiescence in RA patients without anti-rheumatic drugs [152]. Similarly, expression of FcγRIIb or the lack of FcγRIIa or FcR γ-chain on macrophages and DCs may exert negative regulatory roles in TLR4 responses [153–155].
This is why you don’t want off-target effects, eg by accidentally activating FcγRIIa when trying to activate FcγRIIb signalling.
You risk initiating a positive feedback loop:
In human neutrophils, LPS/TLR4 triggers increased expression of FcγRIIa. FcγRIIa and immune complexes also can activate TLR4 signaling cascades in LPS-primed neutrophils [156]. The molecular mechanisms for this crosstalk are not fully understood, but it is Btk dependent [156].
The review “The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease” by Anania et al. (2019)8 adds further to our understanding:
The levels of FcγRIIB expression are influenced by cytokine exposure. Cytokines such as IL-10, IL-6, and dexamethasone increase expression of FcγRIIB, while TNF-α, C5a and IFN-γ inhibit expression (18–20).
On cellular responses:
Follicular dendritic cells (FDCs) originate in the primary follicles and the germinal centres of secondary and tertiary lymphoid organs (SLOs and TLOs)9.
This is probably why IgG4 Abs are persistent after exposure to engineered mRNA agents:
FcγRIIB is upregulated after antigen stimulation via immune complexes on follicular DCs (FDCs) (98). FDCs retain immune complexes and recycle them periodically to their plasma membrane, a process believed to be important in development of B cell immune cell memory (99).
The presentation of immune complexes by activated FDCs expressing FcγRIIB provides antigens to B cells in a highly immunogenic form by multimerising the antigens, thus extensively crosslinking multiple BCRs, minimizing B cell FcγRIIB ITIM-mediated inhibition and providing co-stimulatory signals (100).
Monocyte-derived dendritic cells (moDCs) that were treated with IFNγ to upregulate their activating FcγRs (FcγRI and FcγRIIA) had increased IgG-mediated cellular maturation, while moDCs treated with anti-inflammatory concentrations of soluble monomeric IgG (IVIg) to increase FcγRIIB expression had decreased cellular maturation (18).
Anti-inflammatory cytokines with known involvement with IgG4 class switching lead to enhanced FcγRIIB expression, whilst preventing expression of proinflammatory cytokines by IgG:
Similarly, monocytes with increased expression of activating FcγRs over FcγRIIB as induced by IFNγ or TNFα had enhanced IgG-triggered cytokine production, while monocytes with enhanced FcγRIIB expression by IL-4 and IL-10 prevented IgG-triggered cytokine production (103).
Furthermore, FcγRIIB−/− mouse macrophages developed robust inflammatory responses after exposure to subthreshold concentrations of immune complexes that failed to induce responses in FcγRIIB-expressing cells, demonstrating a role of FcγRIIB in setting a “threshold” for cellular activation (104).
On the outsized effects of IgG4 relative to IgG1 and IgG3:
… FcγRIIB binds IgG4 but not IgG2 and moreover, binds IgG1 and IgG3 an approximately 10-fold lower affinity than the activating FcγRIIA.
You don’t want this, but engineered mRNA-mediated IgG4 is hitting the immune response emergency stop button:

This is consistent with its powerful physiological inhibitory function as IgG binding affinities equal to or higher than the activating receptors might otherwise prevent pro-inflammatory responses that are necessary in resisting infection. Not surprisingly, FcγRIIC has the same IgG binding properties at [as] FcγRIIB (6).
A mutation of the FcγRIIb gene which leads to less immunosuppression is naturally selected for in malaria-exposed areas, as it confers increased survivability.
This also highlights the potentially disastrous consequences of inducing IgG4 class switching at population-level by grossly negligent (or deliberate) mass immunisation programs:
The hypo-functional FcγRIIB-Thr232 variant is enriched in populations from malaria endemic areas. This suggests that reduced FcγRIIB modulation of responses and a consequential enhancement of B cell and inflammatory cell activation confers a survival advantage in these populations (132, 163).
Indeed, enhanced activatory FcR responses including increased phagocytic capacity and TNF production by innate cells and enhanced B cell responses is evident by elevated malaria specific antibody titers (164).
Returning to our last Substack:
Cancer
The roles of FcγR in cancer relate largely to the harnessing of antibody-dependant effector functions such as ADCC or ADCP by therapeutic mAbs during the treatment [reviewed in (2, 139)]. However, it also appears that mAb therapy may also have long term therapeutic benefits.
You need to activate FcγRIIA, but not FcγRIIb, to fight tumours:
Studies on DCs indicate that FcγRIIA activation is necessary and sufficient to induce a strong T cell anti-tumor cellular immunity inducing long term anti-tumor vaccine-like or “vaccinal effects” in humanized mice (48).
Engagement of FcγRIIA induced DC maturation and up-regulation of costimulatory molecules, priming them for optimal antigen presentation and cross-presentation, thus stimulating long-term anti-tumor T cell memory (48).
Conversely, the inhibitory role of FcγRIIB may be disadvantageous to antibody-based therapies and other immune stimulating therapies. Thus, blocking inhibitory function of FcγRIIB on effector cells or antigen presenting cells such as DCs might be a strategy to enhance anti-tumor immune responses during immunotherapy (18, 182, 183).
It’s complicated
Engineered mRNA products were rolled out whilst the underlying mechanisms of immune activation and suppression were not fully understood. They still aren’t.

FcγRIIb can also aggregate together to form complexes, leading to apoptosis of the B cell. In lymph node germinal centres this can have the effect of negatively selecting B cells with BCRs for specific antigens10:
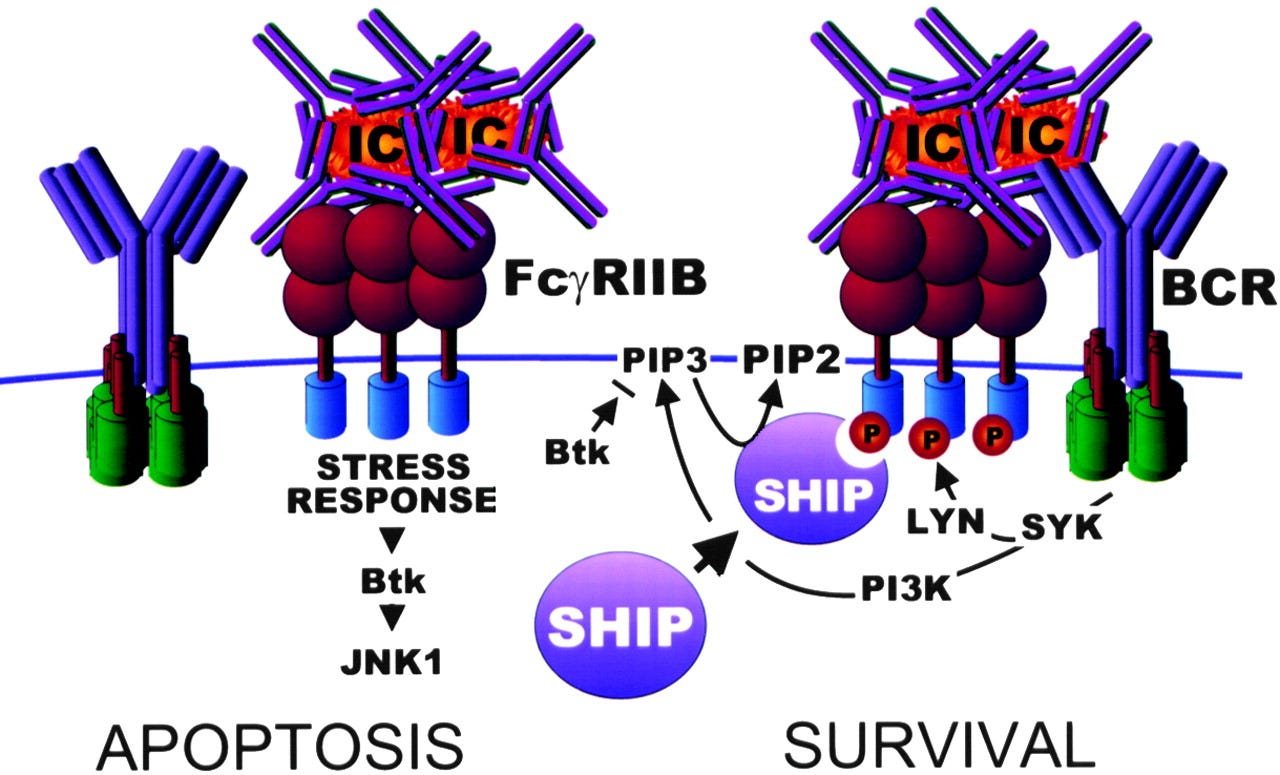
Getahun & Cambier (2016) go into greater depth, and highlight areas where there are gaps in our knowledge.
From “Of ITIMs, ITAMs and ITAMis, revisiting Immunoglobulin Fc Receptor signaling”11:
… The signaling pathway by which FcγRIIb homotypic-aggregation induces apoptosis is still unclear. This pathway is independent of the ITIM, SHIP-1 and Src family kinases, but FcγRIIb is phosphorylated and, though their exact role is unclear, the c-Abl family kinase pathway members, c-Abl and Arg were shown to be of importance (40).
Pre-B cells respond to FcγRIIb aggregation by undergoing apoptosis, but also display impaired cell migration due to SHIP-1 activation. How SHIP-1 becomes activated is unclear because in these cells FcγRIIb does not seem to become phosphorylated (41).
… Finally, it has been shown that by a poorly understood mechanism involving actin depolymerization, FcαRI-SHP-1 complexes translocate into lipid rafts with activating receptors, possibly enabling SHP-1 trans-dephosphorylation of neighboring receptors (72).
This non-covalent co-localization would allow SHP-1 to have a wider reaching inhibitory potential, much like the broader range of SHIP-1 (73, 74). This phenomenon is not unique to FcαRI, similar inhibitory activity has been observed in the case of FcγRIIa (75).
The ITAM FcγRIIa may also be inhibitory if stimulated with intravenous IgG (IVIG) or by F(ab')2 fragments. These sorts of mechanisms help to explain why some mAB treatments have low efficacy with some patients, or why efficacy wanes.
F(ab')2 fragment antibodies are generated by pepsin digestion of whole IgG antibodies (See below IgG structure) to remove most of the Fc region while leaving intact some of the hinge region. F(ab')2 fragments have two antigen-binding F(ab) portions linked together by disulfide bonds, and therefore are divalent with a molecular weight of about 110 kDa.
 folds into a variable domain (VL) and a constant domain (CL) whereas the heavy chain is composed of one variable domain (VH) and three (IgG and IgA or four constant domains (IgE).")
From “Immunoglobulin F(ab) and F(ab')2 fragments“
Stimulation with IVIG or F(ab’)2 anti-FcγRIIa resulted in inhibitory signaling by FcγRIIa independent of FcγRIIb. Low level ITAM phosphorylation reportedly leads to a transient Syk recruitment followed by a stable SHP-1 recruitment.
… It’s complicated, but in the context of stimulation by ligands of differing avidity, ITAMs and ITIMs seem to have enormous potential to tune biological responses. Appropriate surrogates of ligands that activate inhibitory signaling may have great utility in the clinic.
miR-222, tolerance and IgG4-RD
MicroRNA-222 typically acts to promote tumour growth (i.e. it is an oncomiR), and it acts as a biomarker for several cancers. These include breast cancer, liver cancer, pancreatic cancer, prostate cancer, gastric cancer, colorectal cancer, glioma, multiple myeloma, and malignant melanoma12.
From “NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells“13 (2011) by Galardi et al.:
… miRNAs are found to be key players in cancer, where they regulate all main aspects of tumorigenesis and tumor progression, from the first initiating steps to metastasis formation and spreading (2). Those miRNAs whose expression is positively correlated with oncogenesis are often dubbed ‘oncomiRs’.
Among these, miR-221 and miR-222 are a pair of miRNAs encoded in cluster on chromosome X, widely overexpressed in a large variety of human cancers, where they were shown to play their oncogenic roles via the downregulation of several tumor suppressors such as p27, p57, PTEN and many others (3,4).
In our previous work, we demonstrated that miR-221 and miR-222 are overexpressed in prostate carcinoma and in glioblastoma (5–7), and in both tumors they downregulate the cell cycle inhibitor p27 by impairing the translation of its mRNA (6,7).
Montes-Moreno et al. (2011) produced some useful figures showing the association between high levels of oncomiR expression and mortality, and hazard ratios for miR-222 and diffuse large B-cell lymphoma.
From “miRNA expression in diffuse large B-cell lymphoma treated with chemoimmunotherapy”14:




Cancer aside, miR-222 also increases CCL1 and M2b macrophage phenotype counts. Is there a link between CCL1 (a chemokine that attracts immune cells) and IgG4-RD?
“Possible involvement of CCL1-CCR8 interaction in lymphocytic recruitment in IgG4-related sclerosing cholangitis“15 from 2013 by Zen et al. shows that there probably is, at least with the bile duct disease IgG4-related primary sclerosing cholangitis (PSC), and autoimmune pancreatitis (AIP), although they didn’t analyse the miRNA fold changes:
In conjunction with higher expression levels of IL-4 and IL-10, expression values of CCL1 and CCR8 transcripts were significantly higher in IgG4-SC/AIP than in IgG4low PSC (p=0.002) and IgG4high PSC (p=0.023).
CCL1 and CCR8 were also overexpressed in IgG4high PSC than in IgG4low PSC (p=0.023). No difference was seen for CCL17, CCL22, and CCR4.
In situ hybridization revealed CCL1 to be predominantly expressed in the pancreatic duct epithelium, peribiliary glands, and vascular endothelial cells including the ones involved in obliterative phlebitis in IgG4-SC/AIP, in contrast to IgG4high PSC where this chemotactic factor was positive in several infiltrating lymphocytes.
These CCL1-expressing sites were infiltrated by CCR8+ lymphocytes. On immunohistochemistry, GATA3+ Th2 lymphocytes and FOXP3+ Tregs were significantly larger in number in IgG4-SC/AIP, with the GATA3+/T-bet+ cell ratio to be shifted in favour of Th2 in periductal and perivascular areas.
In the context of tolerization to bacterial lipopolysaccharides (LPS, endotoxin), leading to immunosuppression and risk of death from sepsis, Seeley et al. published “Induction of innate immune memory via microRNA targeting of chromatin remodeling factors“16 (2018).
They found that miR-222 upregulation is associated with organ damage and a poor prognosis:
Prolonged exposure to microbial products, e.g. lipopolysaccharide (LPS), can induce a form of innate immune memory that blunts subsequent responses to unrelated pathogens (“LPS tolerance”).
Sepsis, which continues to have a high mortality rate, is a dysregulated, systemic immune response to disseminated infection.
In some patients, this results in a period of immunosuppression (“immunoparalysis”)1 with reduced inflammatory cytokine output2, increased secondary infection3, and increased risk of organ failure and mortality4.
LPS tolerance recapitulates several key features of sepsis-associated immunosuppression5. Although various epigenetic changes have been observed in tolerized macrophages6–8, the molecular basis for tolerance, immunoparalysis, and other forms of innate immune memory has remained unclear.
Here, we performed a screen for tolerance-associated microRNAs (miRNAs) and identified miR-221/222 as regulators of the functional reprogramming of macrophages during LPS tolerization.
Prolonged stimulation with LPS in mice leads to Increased expression of miR-221/222, which regulates brahma-related gene 1 (Brg1) causing transcriptional silencing of a subset of inflammatory genes that depend on SWI/SNF- (SWItch/Sucrose Non-Fermentable) and STAT- (signal transducer and activator of transcription) mediated chromatin remodeling, and promotes tolerance.
In sepsis patients, increased miR-221/222 expression correlates with immunoparalysis and increased organ damage. Hence our results show that specific microRNAs can regulate macrophage tolerization and may serve as biomarkers of immunoparalysis and poor prognosis in sepsis patients.
Linking the 2 previous studies, at least one other paper shows that IgG4-RD, including IgG4-related intrahepatic sclerosing cholangitis, may also lead to sepsis as an outcome17:
We report a case of pre-disseminated intravascular coagulation caused by secondary suppurative inflammation in a patient with immunoglobulin (Ig) G4-related sclerosing cholangitis. The patient was a 78-year-old man in whom a localized stenosis of the intrahepatic bile duct was found without any other bile duct stricture or symptoms. He underwent surgical resection 6 months later for acute severe cholangitis and sepsis caused by bile duct obstruction.
From: “IgG4-related intrahepatic sclerosing cholangitis resulting in sepsis caused by secondary suppurative inflammation: report of a case“ (2012)
I couldn’t find any papers suggesting the reverse though, i.e. that sepsis induces IgG4-RD, perhaps because B cells also get suppressed. However, there is research showing that a treatment for IgG4-RD, Rituximab, is associated with a higher incidence of infection complications, including sepsis.
Rituximab is a monoclonal antibody (MAB) that attaches itself to CD20 proteins on B cells. It causes B cell death via multiple mechanisms18, making it a treatment for some blood cancers (such as Non-Hodgkin’s Lymphoma, NHL) and other autoimmune diseases. The full mode of action is not fully understood.
From: “B cell targeted therapy for immunoglobulin G4-related disease“19 (2021) by Yamamoto:
Numerous patients have been found to require maintenance therapy even after successful induction therapy with rituximab. Omar et al. showed that the risk of relapse tended to be lower with rituximab maintenance therapy than with only rituximab induction therapy after remission [Citation12].
Forty-three patients with autoimmune pancreatitis and IgG4-related sclerosing cholangitis were divided into two groups: those who took rituximab only at the time of the induction therapy and those who continued maintenance therapy with rituximab after remission.
The maintenance group had a significantly lower relapse rate of 11%; however, infection complications (pneumonia, sepsis due to urinary infection, Clostridium difficile colitis, dental abscess, sinusitis, and diverticulitis) were more frequent (20.7%) in the rituximab maintenance group [Citation39].
We have also experienced good results with rituximab induction therapy in patients with repeated relapses. Initially, the clinical symptoms (Figure 2) and serum IgG4 levels improved rapidly in all patients. The use of glucocorticoids was therefore tapered, and rituximab was administered after each relapse (Figure 2); however, the interval between the rituximab administrations gradually became shorter. The patients eventually presented secondary failure [Citation40], which raises the issue of the administration method for the long-term use of rituximab.
At this point, there is no report of attenuated efficacy due to the appearance of anti-chimeric antibody (HACA) in patients treated with rituximab for IgG4-RD, but this phenomenon has been confirmed in rituximab treatment for pediatric systemic lupus erythematosus [Citation41] and Sjögren’s syndrome [Citation42]. HACA may be a possible cause of secondary failure in this disease as well. Optimization of the maintenance therapy with rituximab is therefore essential.
Figure 2. The changes in facial appearance of the patient with immunoglobulin G4-related dacryoadenitis. Rituximab rapidly improved the swollen upper eyelids and reduced the steroid levels, but the cases presented relapse within 3 years.

Rituximab takes out your B cells, and it takes months or even years for levels to normalise after the treatment is ceased:
In systemic lupus erythematosus, rituximab therapy for glucocorticoid-resistant patients resulted in the disappearance of CD19+IgD+CD27− naïve B cells, CD19+IgD−CD27+ class-switched memory B cells, and CD19+IgD−CD27− memory B cells from peripheral blood within 4 weeks.
After that period, naïve B cells recover in approximately 3-9 months and are maintained for 2–6 years.
In patients with systemic lupus erythematosus in remission for a extended period, memory B cells and plasmablasts remain absent for 6 months to 6 years.
Rituximab inhibits the activation and differentiation of T cells, which are normally mediated by memory B cells via co-stimulatory molecules (increase in CD4+ naïve T cells and decrease in CD40L+CD4+ T cells and ICOS+CD4+ T cells), thereby regulating the disease [Citation44].
It has been reported, however, that some patients with systemic lupus erythematosus relapsed after remission with rituximab with no changes in the B-cell lineage but with abnormalities in the T-cell lineage, such as increased CD4+CD45RO+ memory T cells and increased ICOS and CD69 expression in CD4+ T cells [Citation45]. The same phenomenon might occur in IgG4-RD.
Closing remarks
I will stop here as I’ve reached my self-imposed word count limit. I’ve had to cut this review down by 2/3rds as it is, as it was approaching the size of a thesis.
IgG4-RD and its related mechanisms and treatments could easily fill a book, and there are still wide gaps in our knowledge.
Understanding these is the first step in assisting in more accurate, speedy diagnoses, and for the discovery of more effective treatments. Mechanisms of harm of experimental engineered mRNA products only add to the complexity.
Thank you for reading.

DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.

References
Jain, Shilpi, Sanjeev Kumar, Lilin Lai, Susanne Linderman, Ansa A. Malik, Madison L. Ellis, Sucheta Godbole, et al. ‘XBB.1.5 Monovalent Booster Improves Antibody Binding and Neutralization against Emerging SARS-CoV-2 Omicron Variants’. bioRxiv, 5 February 2024. https://doi.org/10.1101/2024.02.03.578771.
Jain S, Kumar S, Lai L, et al. XBB.1.5 monovalent booster improves antibody binding and neutralization against emerging SARS-CoV-2 Omicron variants. Published online February 5, 2024:2024.02.03.578771. doi:10.1101/2024.02.03.578771
Gelderloos AT, Verheul MK, Middelhof I, et al. Repeated COVID-19 mRNA vaccination results in IgG4 class switching and decreased NK cell activation by S1-specific antibodies in older adults. Immunity & Ageing. 2024;21(1):63. doi:10.1186/s12979-024-00466-9
Rubio-Casillas A, Redwan EM, Uversky VN. Does SARS-CoV-2 induce IgG4 synthesis to evade the immune system? Published online August 9, 2023. doi:10.20944/preprints202308.0776.v1
Bianchini R, Karagiannis SN, Jordakieva G, Jensen-Jarolim E. The Role of IgG4 in the Fine Tuning of Tolerance in IgE-Mediated Allergy and Cancer. International Journal of Molecular Sciences. 2020;21(14):5017. doi:10.3390/ijms21145017
Wang L xun, Zhang S xi, Wu H juan, Rong X lu, Guo J. M2b macrophage polarization and its roles in diseases. Journal of Leukocyte Biology. 2019;106(2):345-358. doi:10.1002/JLB.3RU1018-378RR
Li X, Kimberly RP. Targeting the Fc receptor in autoimmune disease. Expert Opin Ther Targets. 2014;18(3):335-350. doi:10.1517/14728222.2014.877891
Anania JC, Chenoweth AM, Wines BD, Hogarth PM. The Human FcγRII (CD32) Family of Leukocyte FcR in Health and Disease. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.00464
Kranich J, Krautler NJ. How Follicular Dendritic Cells Shape the B-Cell Antigenome. Front Immunol. 2016;7. doi:10.3389/fimmu.2016.00225
Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP Recruitment Attenuates FcγRIIB-Induced B Cell Apoptosis. Immunity. 1999;10(6):753-760. doi:10.1016/S1074-7613(00)80074-6
Getahun A, Cambier JC. Of ITIMs, ITAMs and ITAMis, revisiting Immunoglobulin Fc Receptor signaling. Immunol Rev. 2015;268(1):66-73. doi:10.1111/imr.12336
Song J, Ouyang Y, Che J, et al. Potential Value of miR-221/222 as Diagnostic, Prognostic, and Therapeutic Biomarkers for Diseases. Front Immunol. 2017;8. doi:10.3389/fimmu.2017.00056
Galardi S, Mercatelli N, Farace MG, Ciafrè SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res. 2011;39(9):3892-3902. doi:10.1093/nar/gkr006
Montes-Moreno S, Martinez N, Sanchez-Espiridión B, et al. miRNA expression in diffuse large B-cell lymphoma treated with chemoimmunotherapy. Blood. 2011;118(4):1034-1040. doi:10.1182/blood-2010-11-321554
Possible involvement of CCL1-CCR8 interaction in lymphocytic recruitment in IgG4-related sclerosing cholangitis - Journal of Hepatology. Accessed October 8, 2024. https://www.journal-of-hepatology.eu/article/S0168-8278(13)00432-7/abstract
Seeley JJ, Baker R, Mohamed G, et al. Induction of innate immune memory via microRNA targeting of chromatin remodeling factors. Nature. 2018;559(7712):114-119. doi:10.1038/s41586-018-0253-5
Saito M, Shimura T, Hashimoto Y, et al. IgG4-related intrahepatic sclerosing cholangitis resulting in sepsis caused by secondary suppurative inflammation: report of a case. Surg Today. 2013;43(10):1175-1179. doi:10.1007/s00595-012-0448-6
Hanif N, Anwer F. Rituximab. In: StatPearls. StatPearls Publishing; 2024. Accessed October 8, 2024. http://www.ncbi.nlm.nih.gov/books/NBK564374/
Yamamoto M. B cell targeted therapy for immunoglobulin G4-related disease. Immunological Medicine. 2021;44(4):216-222. doi:10.1080/25785826.2021.1886630
Associated outcomes?
Admission trends and waiting lists keep going in one direction, even with making allowances for industrial action.
#Baffled
A&E in England has busiest summer ever with 4.6m visits in two months
The NHS in England has had its busiest summer ever in A&E with 4.6m attendances over the past two months, while 1.5m hospital appointments were rescheduled because of the junior doctors’ strikes, according to the latest figures.
The three busiest months for A&E staff in history have been in 2024, with 77,945 attendances a day in May, 76,469 in June and 74,459 in March.
... Prof Stephen Powis, the NHS national medical director, said: “A&E staff are under significant pressure and the NHS is in the middle of what could be its busiest summer ever, with a total of 4.6m attendances in the last two months alone and 2024 now having seen the three busiest months for A&E on record.
https://www.theguardian.com/society/article/2024/aug/08/ae-in-england-has-busiest-summer-ever-with-46m-visits-in-two-months
If you really want to stop this, produce A5/A4 downloadable leaflet with info in layman terms which these gene "therapy" jabs cause. Have a think on how to do it.