


“Review: N1-methyl-pseudouridine (m1Ψ): Friend or foe of cancer?” - Further reading (Part II)

Reading time:
short story - novelette - novella - novel - PhD thesis - War and Peace - U.S. Tax Code
Also available with the translator, 🇫🇷 🇪🇸 🇩🇪 🇯🇵 etc:
TL;DR: Synthetic mRNA gene therapy: Not only “for them to work, they have to fail”, but to trigger a significant immune response they also need accidental or deliberate adulteration with immunogens too, due to the use of 100% m1ψ. These can trigger type 2 (immune cell associated) interferon (IFN) pathways, but type 1 signalling remains suppressed for at least as long as the transfected mRNA persists in the cell, regardless of what protein is being expressed. This is a problem as “interferon beta (IFN-β) can be produced by all nucleated cells when they recognize that a virus has invaded them”, as well as damping anti-tumor responses.
Going forward, “Personalised medicine” with cancer vaccines is over hyped, has many challenges, and comes with a huge price tag.
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.

From: “Google's Woke AI Is Hilariously But Frighteningly Broken”:

Contents
“Double-edged effects of interferons on the regulation of cancer-immunity cycle”
“The Innate Immune Signalling Pathways: Turning RIG-I Sensor Activation against Cancer”
How does BNT162b2 signal T cell responses, if not through TLR7?
Introduction
In part I I gave some background to the work of the pharma-sponsored “fact checkers”, the latest cancer stats, why NPI’s were never going to work, and further reading relating to our review paper.
In this Substack, I will review a case study, and do a deep dive into why suppressing type 1 interferon signaling pathways with 100% m1ψ modified mRNA to increase protein expression is a terrible idea.
Rather than a Noble, it’s more worthy of a Darwin Award.

They are still pushing product, and it’s laughable that this passes for “Everything you need to know”, especially as the miss the teeny-tiny detail that it was designed to target a variant that rapidly waned to extinction about a year ago. In other words, it doesn’t work.


Discussion
- A case study
In 2023, Kim et al. reported on a case of interferon signaling suppression in myocarditis via a decreased immune response activity (signal transducer and activator of transcription 1, STAT1) after administration of Pfizer’s BNT162b2 synthetic mRNA gene therapy.
The authors considered the paper to be, at the time, a unique study into the epigenomic immune cell landscape of myocarditis caused by the gene agent.
To understand how the immune system was changed they conducted single-cell ATAC sequencing and single-cell RNA sequencing (scRNA-seq) on a patient with myocarditis at the acute inflammatory stage, and again when in remission.
Chromatin: a “mixture of DNA and proteins that form the chromosomes found in the cells of humans and other higher organisms.”

What is ATAC-Seq?

The assay for transposase-accessible chromatin with sequencing (ATAC-Seq) is a popular method for determining chromatin accessibility across the genome. By sequencing regions of open chromatin, ATAC-Seq can help you uncover how chromatin packaging and other factors affect gene expression.
ATAC-Seq does not require prior knowledge of regulatory elements, making it a powerful epigenetic discovery tool. It has been used to better understand chromatin accessibility, transcription factor binding, and gene regulation in complex diseases, embryonic development, T-cell activation, and cancer.1,2 ATAC-Seq can be performed on bulk cell populations or on single cells at high resolution.
… Additionally, ATAC-Seq can be combined with other methods, such as RNA sequencing, for a multiomic approach to studying gene expression.3
‘ATAC-Seq allows you to ask questions about the epigenetic variability in complex or rare tissues and epigenomic landscape in populations of cells that haven’t been observable at the genome-wide level before.’
William Greenleaf, PhD
From: “A rapid, sensitive method for profiling accessible chromatin across the genome“
https://emea.illumina.com/techniques/multiomics/epigenetics/atac-seq-chromatin-accessibility.html
Key takes from “Epigenomic landscape exhibits interferon signaling suppression in the patient of myocarditis after BNT162b2 vaccination“ (2023):
Previously, we had reported the peripheral immune landscape of a 59-year-old male patient who had developed severe myocarditis after BNT162b2 vaccination10.
The immune response of patients with myocarditis after BNT162b2 vaccination was analyzed in more detail by comparing vaccinated individuals without side effects, Coronavirus disease 2019 (COVID-19) patients and Kawasaki disease patients at the single-cell level11
The authors called for more attention to be paid to their findings, and I can see why. In brief, although this is counter-intuitive, they found that BNT162b2-induced suppression of interferon gamma was associated with heart inflammation.
This is the first time I’ve seen a potential link between m1Ψ induced suppression of IFN signaling and myocarditis. Correlation doesn’t equal causation though, so we need to see why the authors think this is happening.
Both type 1 and type 2 IFN signaling were suppressed.
TF’s: Transcription factors bind to specific regions of DNA and modulate transcriptional activity. Outcomes depend on interactions with chromatin-derived nucleosomes.
Suppression of type 1 IFN signaling at the acute myocarditis stage
Next, we investigated TFs whose activities were downregulated at the time of acute myocarditis in the cell subpopulations with the highest number of DEGs. In NK cells and CD8 effector cells, IFN regulatory factors (IRFs) showed the highest degree of downregulation in the myocarditis state.
Look at how all the IRF’s in “A” are downregulated:

Suppression of type 2 IFN signaling at the acute myocarditis stage
… Considering IFNγ signaling-recipient cells, decreased motif activity in STAT1 across multiple cell types in myocarditis suggests that the response to IFNγ secreted from cytotoxic NK and CD8 effector cells was downregulated (Fig. 4B, S9C, and D). IRF1, a target gene of the IFNγ signaling pathway, was also downregulated at the myocarditis state (Figs. 4C, and S9E).
This is what should happen:
During viral infection, viral DNA and RNA activate IRF3/IRF7, which activates IFNA/IFNB gene transcription27,28. Secreted IFNα or IFNβ binds to the IFN-α/β receptor on the surface of almost all cell types, leading to the formation of the STAT1/STAT2/IRF9 complex, known as the ISGF3 complex. This complex initiates the transcription of IFN-stimulated genes to stimulate an immune response to eliminate the viral infection.
Note: Severe COVID also decreases IFN production, but due to lymphopenia, T cell exhaustion (including moderately increased PD-1), and the inhibitory effects of viral proteins (nsp1, nsp12, nsp13, nsp14, nsp15, orf6, M protein, and S protein). A feedback process, impaired IFN signalling also leads to increased viral loads and systemic dissemination.
In the vaccine injured, they found that gene modulation due to interferon regulatory factor (IRF) binding to chromatin was impaired, even though TLR4-triggered levels of IRF3 were not reduced:
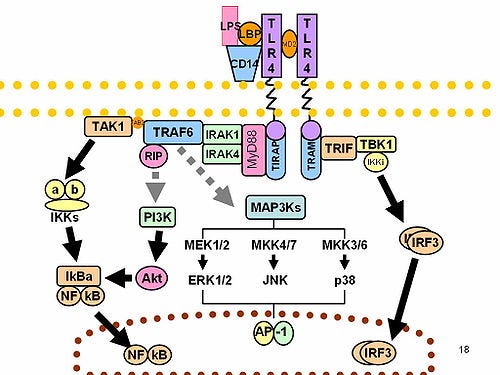
In our datasets, although the gene activity and RNA expression level of IRF3 showed minimal changes (Figs. S7A, and B), the motif activity of IRF3 and IRF7 was decreased at myocarditis state across all cell subpopulations except for pDCs and memory B cells (Figs. 3B–D, and S7C).
When we compared the openness of genomic regions predicted to be the sites of IRF3/7 binding, chromatin accessibility was reduced in myocarditis CD8 effector and cytotoxic NK cells (Fig. S8A, and B).
In addition, we performed Gene Set Variation Analysis (GSVA), which calculates gene set enrichment scores for a sample to validate changes in RNA expression profile in response to Type I IFN30.
A decrease in IFNα/β-related gene expression was primarily observed in T cells. IFNβ had a more profound influence on the immune cell response in myocarditis (Fig. 3G).
They also found that type 2 IFN signaling was impaired in cytotoxic natural killer (NK) cells and CD8 T cells, along with decreased chromatin accessibility. What came first, the myocarditis or impaired IFN signaling?
We first investigated the mRNA expression levels and distribution patterns of IFN-γ. Cytotoxic NK and CD8 T cells showed abundant expression and decreased expression levels at the time of myocarditis, respectively (Figs. 4A, S9A).
In addition, there was a mild decrease in chromatin accessibility with a reduction in mRNA expression level at the region, which Cicero predicts as a cis-regulatory DNA region for IFNG transcription (Fig. S9B).
Does this support the m1Ψ study findings, with an added complication of blunted responses?
Considering IFNγ signaling-recipient cells, decreased motif activity in STAT1 across multiple cell types in myocarditis suggests that the response to IFNγ secreted from cytotoxic NK and CD8 effector cells was downregulated (Fig. 4B, S9C, and D). IRF1, a target gene of the IFNγ signaling pathway, was also downregulated at the myocarditis state (Figs. 4C, and S9E).
The GSVA score analyzed from mRNA expression levels advocates blunted response to IFNγ in CD14 monocyte, cytotoxic NK, CD8 T, Treg, CD4 memory, and CD4 naïve cells (Fig.(Fig.4D4D and E).
Therefore, the immune system of a patient with acute myocarditis after BNT162b2 vaccination exhibited impaired IFN signaling, although aggregated peaks in the regulatory element of pro-inflammatory cytokine were increased (Fig. S10).
As with TLR4 & IRF3, serum IFN increases after BNT162b2. But, as we shall see later, this is not necessarily a balanced, natural IFN pathway with anti-tumour efficacy. Indeed the TLR4/TRIF/IRF3 signaling pathway contributes to tumor progression and metastasis in some cancers.
Considering that BNT162b2 vaccination induces the upregulation of serum IFN levels, the patient with the side effect of vaccination is likely to develop immune responses different from what has been known31.
m1Ψ impaired signaling and gene expression:

Inflammatory cytokines were upregulated, but not interferon signaling:
Integrated analysis of scRNA-seq and scATAC-seq from our results revealed a downregulated IFN signaling pathway and upregulated gene activities of IL-1, IL-6, IL-17, and IL-21. Thus, patients with myocarditis after BNT162b2 vaccination displayed a hallmark of decreased IFN (type I and type III) signaling and increased IL-6 production, which resembles the characteristics of bronchial epithelial cells.
Note the type of interferon that increases in vaxxed healthy adults: Gamma.
After booster vaccination in healthy adults, IFNγ secretion increases in CD8 and CD4 T cells36.
Meta-analysis in COVID-19 patients with or without severe symptoms also reported that patients with severe COVID-19 have a high IL-6/IFNγ ratio37. Therefore, IFN signaling in patients with myocarditis after vaccination appears to be similar to that in patients with severe COVID-19.
In this study, we verified decreased IFN signaling in the NK cells of patients with severe side effects of BNT162b2 vaccination by analyzing epigenomic profiles. In future studies, more attention should be paid to IFN signaling in NK cells in myocarditis.
No competing interests were declared.
“High–temporal resolution profiling reveals distinct immune trajectories following the first and second doses of COVID-19 mRNA vaccines” (2022) by Rinchai et al. investigated interferon responses and differences post-first and post-second dose.
Key takes:
The first vaccine dose elicited modest interferon and adaptive immune responses, which peaked on days 2 and 5, respectively.
The second vaccine dose, in contrast, elicited sharp day 1 interferon, inflammation, and erythroid cell responses, followed by a day 5 plasmablast response.
Both post-first and post-second dose interferon signatures were associated with the subsequent development of antibody responses. Yet, we observed distinct interferon response patterns after each of the doses that may reflect quantitative or qualitative differences in interferon induction.
The timeline was quite detailed, over 9 consecutive days post-vax dose 1 and 2:
In the present study, we endeavored to profile the blood transcriptome of individuals before the administration of the first dose of COVID-19 mRNA vaccines and for the following nine consecutive days. Subjects also collected samples for deep serological profiling at three time points.
The same sampling and profiling schedule was repeated to assess the response to the second dose of the vaccine. To achieve this, we have adopted an ultralow-volume sampling procedure for self-collection and RNA preservation of a few drops of blood (50 μl) collected by a fingerstick (7).
A28 is a CD4+ T cell line:
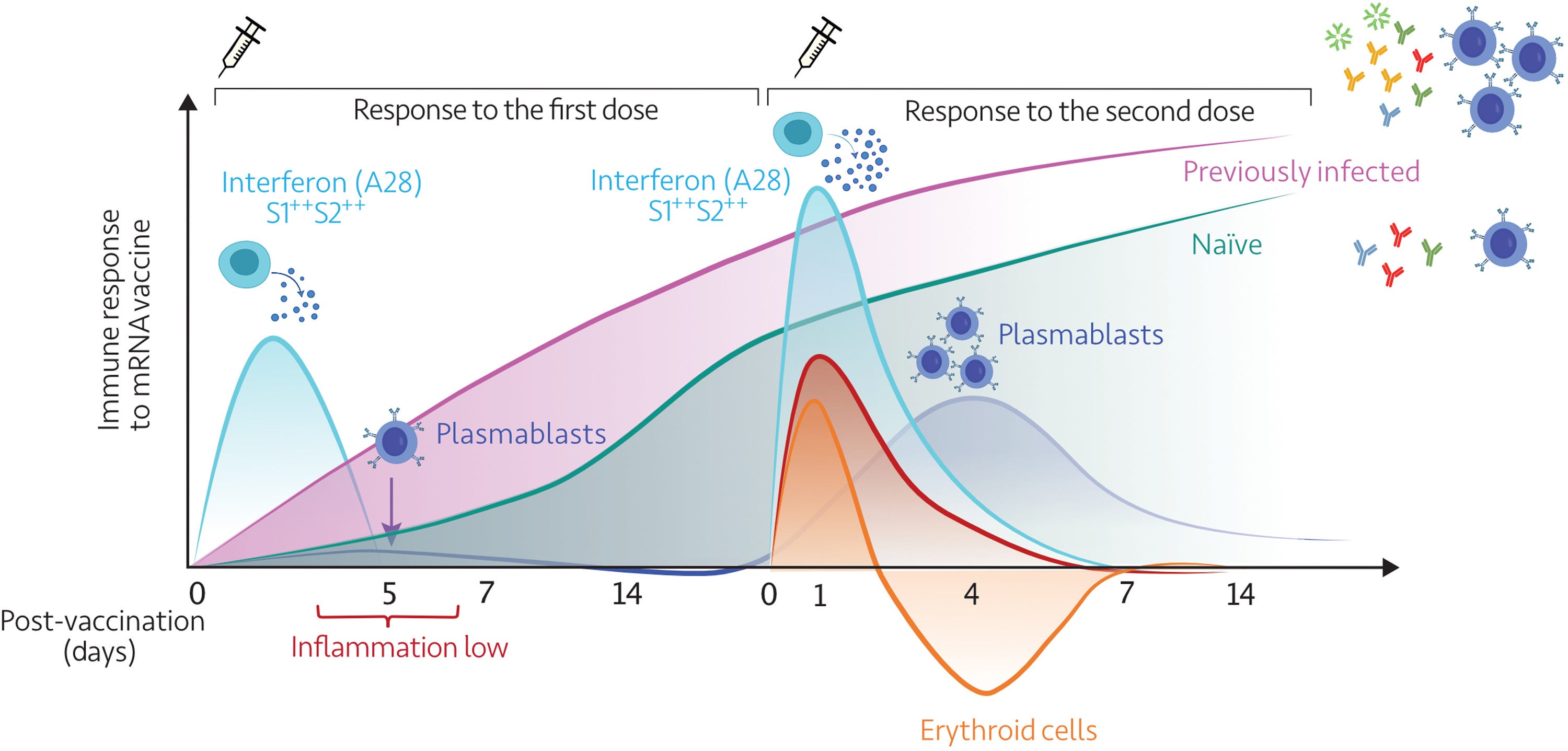
“Immunosuppressive states”, peaking at around day four after the second dose. This is not good. At the time I was trying to warn the vaxxed on Facebook to try to avoid getting infected for a few days after being dosed. I’m not sure they saw the irony in this. The link with cancer also stands out:
In addition to eliminating potential blind spots, high-frequency sampling and profiling also permit the precise resolution of the complex kinetics of a response; for instance, the erythroid cell signature peaks sharply after the second dose and recedes well below baseline over several days before recovering. The trajectory of this signature may be of significance in the context of vaccination, as we recently described its association with immunosuppressive states, such as late-stage cancer and maintenance therapy in liver transplant recipients (28).
Dose 1 was dominated by type 1 interferons, such as INF-α, and dose 2 by type 2 interferon, IFN-γ:
Together, the differences observed after the first and second doses could be attributed to quantitative or qualitative differences, e.g., elicitation by the second dose of vaccine of the release of higher amounts of endogenous interferons or of the release of interferon of a different type (with the post–first dose signature possibly being elicited by type I interferons, as patterns of induction after interferon-β treatment would suggest, and the post–second dose signature possibly being elicited by type II interferon).
The latter thesis is indirectly supported by recent findings in mice that received the Pfizer BioNTech BNT162b2 vaccine and were found to preferentially produce interferon-α in vivo after the first dose of vaccine and interferon-γ after the second dose (44).
It should be, however, noted that while the pattern of induction of A28/S1 and S2 modules might be indicative of a given type of interferon response, our observations would not support the notion of a given set to be exclusively associated with a given type of response (i.e., the analyses of public datasets in fig. S7 show that A28/S1 modules may be more responsive to type I interferons, while the A28/S2 also appears to respond but only to a lesser degree).
Competing interests: D.R.G., outside of this work, reports investigator-initiated grants from Gilead Italia and Pfizer Inc. M.Ba., outside of this work, reports research grants from and/or advisor/consultant and/or speaker/chairman activities for Bayer, Biomerieux, Cidara, Cipla, Gilead, Menarini, MSD, Pfizer, and Shionogi. C.D., outside of this work, reports consultancy/speaker activities for Angelini, MSD, and Novartis. K.S., outside of this work, reports paid consulting for Sanofi, Novo Nordisk, and Roche. All other authors declare that they have no competing interests.
“Toll-like receptor 7 (TLR7) signaling predominantly regulates production of type I interferons (IFNs)”
As our review discussed and the previous study found, interferon responses were dominated by type 2 interferon signaling.
Both types of interferon may have anti-cancer or pro-tumor effects. Type 2 interferons have IFN-γ as their sole representative.

From minireview “Type II Interferon: The "Double Agent" in the Tumor Microenvironment”:
Anti-tumor Mechanism of IFN-γ
The anti-tumor function of IFN-γ involves multiple steps. IFN-γ enhances tumor cell apoptosis by upregulating the expression of caspase-1, -3, and -8 and increasing the secretion of FAS and its ligand and TNF-related apoptosis-inducing ligand. IFN-γ inhibits angiogenesis, damages endothelial cell proliferation and survival, and induces ischemia in the tumor matrix. IFN-γ promotes the recruitment of immune cells to TME through transcriptional regulation of T cells, NK cells, monocytes, dendritic cells, and CXCL9, CXCL10, and CXCL11 on cancer cells, as well as their homologous receptors, thereby limiting tumor growth. TH1 cells producing IFN-γ decrease IFNGR2 expression after differentiation, which enhances their survival rate and anti-tumor efficacy. IFN-γ also plays a role in the tumor-killing function of macrophages.
We discussed 100% m1Ψ associated PD-L1+ T cell exhaustion in our paper:
Pro-tumor Mechanism of IFN-γ
The pro-tumor function of IFN-γ is also achieved through different cellular responses. In mouse models with a low tumor burden, CTLs overexpress IFNGR, leading to the death of activated CTL cells and promoting tumor growth. IFN-γ can upregulate PDL1 expression in various cells, inhibit Th1 cell differentiation, and impair effective tumor immunity. IFN-γ also promotes apoptosis by reducing the expression of Bcl-2, upregulating Fas and Fasl, and generating a harmful oxidative environment. Tumor cells that lose the IFN-γ receptor or a JAK/STAT signaling pathway component undergo immune evasion, inhibiting the immune response mediated by cytotoxic T cells and NK cells.
IFN-γ effects on tumors still aren’t fully understood, yet we mass administered regardless:
More than 50 years have passed since the discovery of IFN-γ, yet the intricate mechanisms of this multifunctional cytokine in tumors are still not fully understood, necessitating further research. Immunotherapy-induced IFN-γ can produce contradictory anti-tumor or pro-tumor effects. Future investigations must employ new methods to discern the pro-inflammatory and anti-inflammatory functions of IFN-γ. This understanding will be crucial in designing improved therapeutic drugs that enhance their anti-tumor capabilities while preventing immune evasion.
Another study, “Double-edged effects of interferons on the regulation of cancer-immunity cycle” from 2021 by Zhang et al. further supports the importance of type 1 interferons in cancer immunity:
Although IFNs were originally identified as potent anti-viral factors, they were also recognized to regulate immune responses and inhibit cancers. Before IFNs were purified, scientists employed unpurified interferon to treat various types of cancer in mice and patients.5
Since interferon possesses anti-viral effects and virus infection is associated with some malignancies (for example, Rous sarcoma virus cause Rous sarcoma), researchers began to use IFNs to treat virus-induced tumors in animal models in the mid-1960s and observed therapeutic effects on these tumors.3,5
The first clinical trial using IFN to treat cancer was initiated in 1971 in osteosarcoma,6 and now IFNs have been used to treat various types of cancer in the clinic, including melanoma, hairy cell leukemia, and renal cell carcinoma.7
However, IFN treatment has minimal or even adverse effects in some instances,8 which suggests that IFNs play a complicated role in the cancer immune response.
IFN-γ is the only member of type II IFN and binds to the IFN-γ receptor composed of IFNGR1 and IFNGR2.
You can see why suppression of type 1 IFNs is much worse than suppressing type 2 IFNs. Many stimuli can trigger them, most cell types can express them. It’s an immune (and therefore anti-cancer) response that is not restricted to immune cells:
There are many intracellular and extracellular stimuli that trigger the production of type I IFNs. Typically, upon infection with microbes or exposure to damaged cellular components, danger-associated molecular patterns (DAMPs) can be recognized by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), cyclic GMP-AMP synthase (cGAS), MDA-5, DAI, RIG-I like receptors, and DDX41in the cell membrane or cytoplasm.
“IFNβ may be considered the “primary” IFN-I because it is expressed by all nucleated cells and may be expressed in isolation of all other IFN-I (except IFNα1…”
After recognizing and binding with DAMPs, these PRRs are activated and interact with adaptor proteins and activate kinases to phosphorylate NF-ĸB, IFN regulatory factor 3 (IRF3), and activating protein 1 (AP-1), which translocate into the nucleus and induce the expression of IFN-β in most cell types in the body28 (Figure 1).
Rather than TLR7 detecting viral mRNA and triggering IFN-β, infection is well underway before IFN-γ is triggered:
In contrast to type I and type III IFNs, the resource of IFN-γ is restricted in immune cells, such as T cells, B cells, natural killer (NK) cells, natural killer T (NKT) cells, DCs, and macrophages.30–37 Many cytokines, such as IL-1, IL-2, IL-12, IL-15, IL-18, IL-21, IL-23, IL-27, IFN-α/β and TNF-α, can induce IFN-γ secretion in various types of immune cells.38

Unsurprisingly, cancer therapies tend to focus on type 1 IFN signaling:
The implication of IFNs in cancer therapy
Given that the induction of IFNs is largely triggered by multiple DAMPs and the activation of IFN signaling pathway exhibits cell intrinsic (anti-proliferation and inducing cell death) and extrinsic (immunomodulation) anti-cancer activity, it seems reasonable to conclude that IFNs play a critical role in the successes of conventional cancer therapeutic strategies.
Indeed, IFNs alone or strategies of stimulating IFN production, and combined using IFNs and other cancer therapies have been demonstrated to be effective to treat various malignancies. Additionally, it has also been revealed that deficiency of IFN signaling is one of the most important reasons for the resistance or failure of common cancer therapeutic strategies.
Firstly, the efficient type I IFN signaling was recognized as a footstone closely related to the success of conventional cancer therapeutic strategies, such as chemotherapy, radiotherapy and immunotherapy.42 Sistigu et al. demonstrated that cancericidal effects of anthracyclines rely on cancer cell autonomously producing type I IFN induced by the activation of toll-like receptor 3 (TLR3).43
Chemotherapeutic drug cyclophosphamide was revealed to modulate the transcriptional prolife of peripheral blood mononuclear cells (PBMCs) in patients with hematologic malignancies and induce a type I IFN associated sterile inflammation, which contributes to cancer cell elimination.44–46
We discussed radiotherapy resistance too:
Apart from chemotherapy, the efficacy of radiotherapy was also highly entwined with the activation of type I IFN signaling.47,48 Both studies in mouse models of melanoma and colorectal carcinoma indicated that radiotherapy induces the production of type I IFN in myeloid cells and thus attributes to the generation of tumor infiltrating DC with enhanced ability to prime T cells.47,48
Additionally, both type I IFN (IFN-α and IFN-β) and type II IFN have been reported to enhance the efficacy of anti-PD1 or anti-PD-L1 in various cancer types, such as melanoma and pancreatic cancer.13–17
Turning down your PRRs also impairs the efficacy of your combined treatment programs:
Secondly, various IFN stimulating strategies based on targeting PRRs have been developed to treat cancer, and accumulating evidence indicate that PRR agonists synergize with other therapy approaches and attribute to a better therapeutic efficacy.
STING: Stimulator of interferon genes.
Deng et al. found that the administration of STING agonist (2ʹ3’ cGAMP, 10μg) synergized with radiation (20 Gy) and significantly boost anti-cancer immune response in murine colon cancer bearing mouse models.48
Ghaffari et al. also showed that STING agonist (2′3′-c-di-AM, 4 mg/kg i.p.) combined with anti-PD-1 antibody, greatly promotes IFN response and the expression of MHC class II genes and subsequently amplifies the therapeutic efficacy of carboplatin in the murine model of high-grade serous ovarian cancer.49
There are many more studies cited in the full paper.
Thirdly, accumulating evidence indicated that deficiency of IFN signaling is one of the most important reasons for the immune dysfunction and even the resistance or failure of common cancer therapeutic strategies.
Another mouse-IFN-melanoma study:
For example, the efficacy of immune checkpoint blockade therapy was significantly reduced on STING knockout mice bearing B16-SIY melanoma, because the loss of STING signaling impaired the tumor-cell-derived DNA triggered production of type I IFN and thus failed to activate DCs.53
As per the study by Sittplangkoon et al.:
Similarly, it was showen [sic] that the downstream targets of IFN-γ were downregulated in different melanoma cell lines with the disappointing response to immunotherapies, suggesting downregulation of IFN-γ signaling is common in melanoma and potentially predicts the response to immunotherapy.56
In summary, it is a promising strategy to use IFNs alone or combined with therapeutic strategies to treat cancer. Preclinical studies have indicated that IFNs are competent in provoking cancer immunity in different cancer types…
Lurescia et al. included some excellent schematics showing the mechanisms involved in RIG-I triggered immune responses to tumor cells. As we discussed in our review, systemic transfection of 100% m1Ψ-mRNA may almost totally “dampen” this type of response.
Key takes from “The Innate Immune Signalling Pathways: Turning RIG-I Sensor Activation against Cancer” (2020).
Figure 1 shows how RIG-I ligands can be detected by the RIG-I PRR, leading to tumor cell apoptosis via a mitochondrial protein called MAVS and through the type 1 IFN pathway.
RIG I may be activated by small nuclear RNA (snRNA) end endogenous retroviral elements (ERVs) that can leak from the nucleus of a tumor cell during programmed cell death, in a virtuous positive feedback loop.
Other ligands can include products of radiotherapy and hypomethylation agents such as 5-azacytidine. These may be given alongside immune checkpoint inhibitors for synergistic benefits:




It would be instructive to compare cancer incidence trends by type since 2021:
Intrinsic/mitochondrial apoptosis was reported to occur by RIG-I activation in melanoma cells, lung cancer, cervical cancer, myeloid leukaemia, colon and prostate carcinoma [51,54,58,59,60,61].
This apoptotic pathway is regulated by the proapoptotic BCL2 family members (e.g., NOXA and PUMA) and results in permeabilization of the mitochondrial outer membrane, with subsequent leakage of apoptosis-inducing proteins, such as cytochrome c, out of the mitochondria.
This, in turn, activates the proapoptotic protein APAF-1, which induces the cleavage of the proenzyme of caspase-9 into the active form. The caspase cascade ultimately results in the proteolysis of intra-cellular proteins and cellular destruction (Figure 3a).
The extrinsic tumour cell death was reported to occur in melanoma, prostate and breast cancers in response to RIG-I signalling [50,55,56]. The extrinsic pathway involves transduction of the apoptotic signal after the engagement of a death receptor by its cognate death ligand. The resulting activation of initiator caspase-8 or -10 then directly activates the effector caspases-3, -6, and -7, effectively executing cellular apoptosis (Figure 3b).
In this regard, upregulation of TNF-related apoptosis inducing ligand (TRAIL), caspases 8 and 10 was detected in prostate cancer and breast cancer cells [50,55]. RIG-I signalling can also induce pyroptosis, an immunogenic form of gasdermin D-mediated necrosis, in breast cancer cells [50].
“Nice looking NK action against tumor cells you got there. Be a shame if something happened to it”:
Very recently, the study by Dassler-Plenker et al. also demonstrated functional RIG-I expression in naïve human NK cells and their direct activation upon RIG-I ligand (3pRNA) transfection.
NK stimulation resulted in increased surface expression of membrane-bound TRAIL, which in turn initiated the extrinsic apoptosis in melanoma cells. TRAIL-dependent killing is induced not only in human allogeneic but also in autologous human leukocyte antigen (HLA) class I-positive and negative melanoma cells [56].
Strikingly, NK cells showed their TRAIL-mediated cytotoxic function only toward cancer cells, sparing healthy normal cells. Non-malignant cells are likely protected by the expression of the decoy receptors TRAIL-R3 and TRAIL-R4, which lack a functional cytoplasmic death domain and fail to induce cell death.
Malignant cells are actually highly sensitive to RIG-I-dependent proapoptotic signals. siRNA-mediated BCL2 silencing as well as TRAIL-dependent NK cells activation provoked massive cell death in cancer cells, while sparing healthy normal cells due to the expression of Bcl-xL, which counteracts RIG-I mediated apoptosis [54], or TRAIL-R3 and TRAIL-R4, which antagonise TRAIL-death signal [56].
They knew that m1Ψ suppresses RIG-I signaling, but not what effects it might have on breast cancer.
“Antimetastatic” means that RIG-I has tumor-suppressing properties:
Despite being the second leading cause of cancer death among women, little is known about RIG-I signalling in breast cancer. Previous reports identified RIG-I as belonging to a pro-inflammatory and antimetastatic gene expression signatures in MDA-MB-435 human breast cancer cells [78,79].
You need both pathways for optimal anti-tumor responses.
TME: Tumor microenvironment.
The RIG-I adapter MAVS and DC-intrinsic type I IFN signalling are functionally required for optimal T-cell expansion and cytolytic activity. These findings validate previous studies demonstrating that IFN I signalling is critical for DCs recruitment in the TME, cross-priming of naïve cytotoxic T-cells, and tumour rejection [12,80,81].
It doesn’t get much clearer than this paper from the following year by Guo et al.
We see a pattern of tumor suppressor downregulation by synthetic mRNA gene agents, and we can add RIG-I to the list:
Key takes from “RIG-I acts as a tumor suppressor in melanoma via regulating the activation of the MKK/p38MAPK signaling pathway“ (2022)
Studies have indicated that RIG-I may act as a tumor suppressor and participate in the tumorigenesis of some malignant diseases. However, RIG-I induces distinct cellular responses via different downstream signaling pathways depending on the cell type.
To investigate the biological function and underlying molecular mechanism of RIG-I in the tumorigenesis of melanoma, we constructed RIG-I knockout, RIG-I-overexpressing B16-F10 and RIG-I knockdown A375 melanoma cell lines, and analyzed the RIG-I-mediated change in the biological behavior of tumor cells in spontaneous and poly (I:C)-induced RIG-I activation.
Our data showed that RIG-I promotes apoptosis and inhibits proliferation by G1 phase cell cycle arrest in the melanoma cell lines. Mechanistically, RIG-I induced the phosphorylation of p38 MAPK and MAPK kinases MKK3 and MKK4. In conclusion, the current study demonstrated that RIG-I suppressed the development of melanoma by regulating the activity of the MKK/p38 MAPK signaling pathway, which is relevant to research on novel therapeutic targets for this malignant disease.
This is awkward and mirrors the findings of the Sittplangkoon et al. study. Knocking out RIG-I allows melanoma cells to proliferate, despite the presence of an immunostimulant.
The effect of RIG-I deficiency on the growth of B16-F10 cells was first evaluated by CCK-8 assay. As shown in Fig. 2A, the relative cell viability of B16-F10 cells was significantly increased in the RIG-I knockout group compared with that in the control group, both in the spontaneous and poly (I: C)-stimulated state.
The results showed that target deletion of RIG-I resulted in a decreased percentage of cells in the G0/G1 phase (spontaneous: 64.97 ± 0.15 vs. 68.17 ± 0.85%, P < 0.001; poly (I: C)-stimulation: 68.6 ± 0.50 vs. 80.93 ± 0.32%, P < 0.0001) and an increased percentage of cells in the S phase (spontaneous: 29.23 ± 0.46 vs. 25.3 ± 1.05%, P < 0.01; poly (I: C)-stimulation: 22.30 ± 1.22% vs. 9.72 ± 0.52%, P < 0.0001) (Fig. 2B, C).
siRNA: Special interfering RNA. In this case, it stops RIG-I from being a tumor suppressor:
Moreover, RIG-I siRNA-transfected A375 human melanoma line showed significantly increased cell viability (Fig. 3A). The results of BrdU incorporation assay also showed that RIG-I knockdown resulted in a decreased percentage of cells in the G0/G1 phase, and increased percentage of cells in the G2/M and S phase (Fig. 3B, C). These data suggested that RIG-I deficiency promotes melanoma cell proliferation.
KO: RIG-I knockout.
Lipo: Lipofectamine 3000, a lipid based transfection agent.
Poly(I:C): Polyinosinic:polycytidylic acid, an immunostimulant.
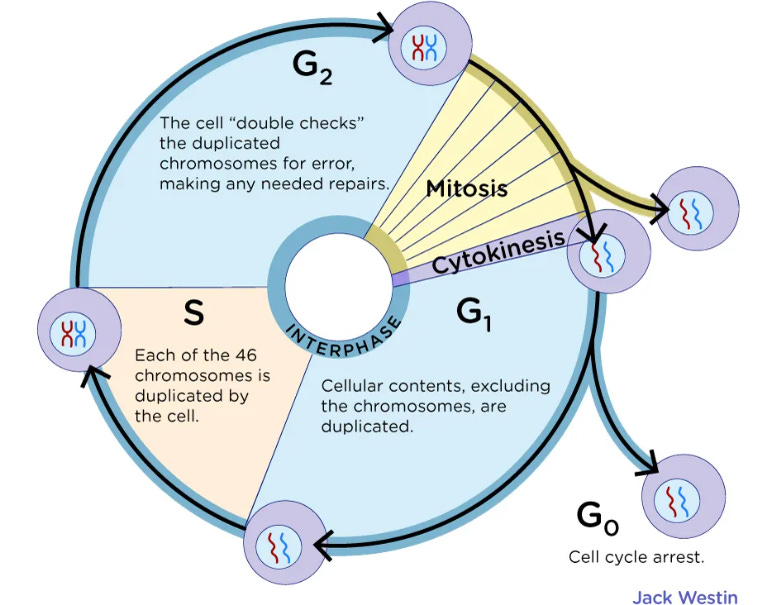
G0: Cell cycle arrest. A goal of anti-cancer drugs and an effect of some tumor suppressors, prior to apoptosis.
G1 phase is where a cell grows and accumulates the building blocks of chromosomal DNA and the associated proteins as well as sufficient energy reserves to complete the task of replicating each chromosome in the nucleus.
S phase is a stage in cell division of DNA synthesis where the genetic content is replicated. RIG-I KO is associated with increased melanoma DNA synthesis
G2 phase checkpoints monitor the integrity of DNA and the accuracy of DNA replication, and the M phase checkpoints ensure correct chromosomal segregation and alignment.

You might call this the “kill shot”:
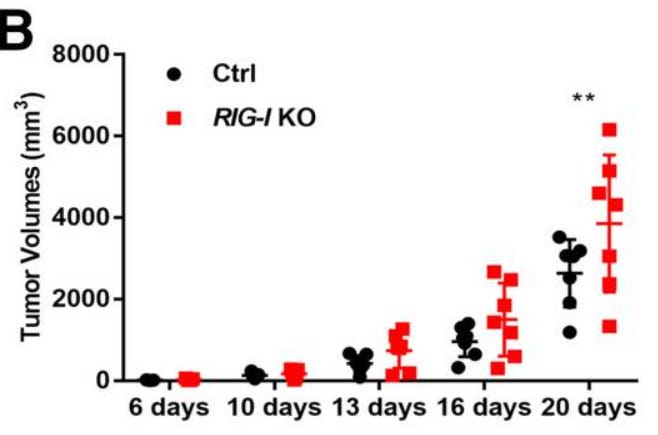
After it was determined that RIG-I promoted apoptosis and suppressed the proliferation of melanoma cells in vitro, the effects of RIG-I deletion on xenograft tumors in nude mice were investigated.
As shown in (Fig. 7A, B), the tumors in mice injected with RIG-I-KO cells showed an obviously faster growth rate than those in the controls. The difference in tumor volume was significant 20 days after subcutaneous injection (P < 0.01).
The proliferative ability of the tumor xenografts was evaluated via histopathological staining for Ki67. The results showed that target deletion of RIG-I could induce proliferation in the tumor xenografts of B16-F10 cells (60.97 ± 13.56 vs. 35.96 ± 7.22%, P < 0.001) (Fig. 7C).
Subsequently, apoptosis in the tumor tissue was examined using a TUNEL assay. The percentage of apoptotic cells in RIG-I-KO tumor xenografts (0.68 ± 0.32%) was significantly decreased compared with that in the control sample (1.61 ± 0.93%) (Fig. 7D). These results demonstrated that RIG-I deletion facilitated tumorigenesis in a xenograft tumor model in vivo.

Extracts
RIG-I knockout induced proliferation in the tumor xenografts:


… accumulating evidence suggests that RIG-I is a multifunctional protein that functions far beyond being a pattern recognition receptor and participates in various biological processes, such as cell proliferation, apoptosis, aging, inflammation, detection of bacterial pathogens and even recognition of self mRNAs [16].
I thought the idea was to promote it, not suppress it:
In the past several years, there has been increased interest in the potential role of RIG-I in the development and immune therapy of many malignant diseases.
Although more precise studies are needed to verify the molecular regulatory mechanism of RIG-I in the pathogenesis of melanoma, to the best of our knowledge, the current study is the first to demonstrate a possible role of RIG-I-mediated activation of the MKK/p38 signaling cascade in the proliferation and apoptosis of melanoma cells.
Our findings might deepen the understanding of the antitumor function of RIG-I in melanoma, which could be useful in elucidating the pathogenesis of and developing new therapeutic strategies for this malignant disease.

Making synthetic mRNA gene therapy treatment de facto mandatory for cancer treatment was always reckless or grossly negligent bordering on insane, given the lack of long-term safety or efficacy data.
Even by as early as July 2020, long before the vaccine rollout, we knew from the CDC that the infection fatality rate (IFR) for COVID-19 averaged around 0.03%, or that of flu.
The argument that cancer patients were at greater risk due to immunosuppression also falls flat. Without a robust immune system, vaccination fails too, and can make things worse due to inducing exhaustion or misdirecting responses away from the tumor site.
Alternatives like ivermectin were available, and “no-action” was always an option - the precautionary principle.
Well done to the 10 of 522 cancer patients (1.9%), who had more sense than the so-called “experts” here. And this isn’t with the benefit of hindsight, many of us were horrified by this level of coercion at the time, and the reports of myocarditis and sudden death that were filtering through by early 2021.
As for “… the duty to ‘protect each other’”, the jabs never stopped transmission. It was turtles all the way down:
Background
At the start of 2021, oncologists lacked the necessary scientific knowledge to adapt their clinical practices optimally when faced with cancer patients refusing or reluctant to be vaccinated against COVID-19, despite the marked vulnerability of these patients to severe, and even fatal forms of this new viral infectious disease. Oncologists at Foch Hospital were confronted with this phenomenon, which was observed worldwide, in both the general population and the population of cancer patients.
Methods
Between April and November 2021, the Ethics and Oncology Departments of Foch Hospital decided to investigate this subject, through an empirical and interdisciplinary study in bioethics. Our scientific objective was to try to identify and resolve the principal bio-ethical issues, with a view to improving clinical practices in oncology during future major pandemics of this kind, from a highly specific bio-ethical standpoint (= quality of life/survival). We used a mainly qualitative methodological approach based on questionnaires and interviews.
Results
In April 2021, 29 cancer patients refused or were reluctant to be vaccinated (5.6%; 29/522). Seventeen of these patients said that making vaccination mandatory would have helped them to accept vaccination. In October 2021, only 10 cancer patients continued to maintain their refusal (1.9%; 10/522). One of the main reasons for the decrease in refusals was probably the introduction of the “pass sanitaire” (health pass) in July 2021, which rendered vaccination indispensable for many activities. However, even this was not sufficient to convince these 10 cancer patients.
Conclusion
We identified a key bio-ethical issue, which we then tried to resolve: vaccination policy. We characterized a major tension between “the recommendation of anti-COVID-19 vaccination” (a new clinical practice) and “free will” (a moral value), and the duty to “protect each other” (a moral standard). Mandatory vaccination, at least in France, could resolve this tension, with positive effects on quality of life (i.e. happiness), or survival, in cancer patients initially refusing or reluctant to be vaccinated, but only if collective and individual scales are clearly distinguished.
Keywords: Pandemic, COVID-19, Cancer, Vaccine, Oncology, Bioethics, Hospital, France
How does BNT162b2 signal T cell responses, if not through TLR7?
Although impaired, we know experimentally that there is a type I IFN response after the first shot, and a type II response after the second. But if not through 100% m1Ψ modified mRNA to TLR7, then how is it being signalled to the immune system?
In 2022, Li et al. conducted a study using knockout mice to try to answer this question. They found that the RIG-I-like receptor to double-stranded RNA (dsRNA), melanoma differentiation-associated protein 5 (MDA5), was likely the key PRR involved.
Key takes from “Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine“ (2022). I first discussed this paper in Part I, in the context of dendritic cell and macrophage transfection:
Analysis of knockout mice revealed that induction of antibody and T cell responses to BNT162b2 was not dependent on signaling via Toll-like receptors 2, 3, 4, 5 and 7 nor inflammasome activation, nor the necroptosis or pyroptosis cell death pathways. Rather, the CD8+ T cell response induced by BNT162b2 was dependent on type I interferon-dependent MDA5 signaling. These results provide insights into the molecular mechanisms by which the BNT162b2 vaccine stimulates immune responses.
Kariko et al. found that the m1Ψ-modification on RNA dampens the inflammatory response mediated by TLR3, TLR7, or RIG-I, and increases translational efficiency and biological stability 3, 4. mRNA vaccines encapsulated in LNPs were also found to induce a high magnitude of germinal center B (GC B) cell and follicular helper T cells (TFH) responses after a single immunization 5, 6, 7, 8.
Mass administration despite their being “major knowledge gaps”, including in relation to tumors or pre-cancerous lesions. No genotoxic studies were ever conducted or requested prior to approval:
Despite our emerging understanding of immune responses to BNT162b2, there are major knowledge gaps on its mechanisms of action. For example, there is a paucity of understanding on the nature of the innate response to BNT162b2 and, in particular, how BNT162b2 is sensed by the host’s innate immune system.
Only for cancer vaccines with zero percent m1Ψ-modified mRNA:
Studies of mRNA vaccines against cancer demonstrate that they can trigger TLR4 14, TLR7 15, or STING 16 signaling pathways, but such knowledge is lacking in the context of BNT162b2.
Our analysis demonstrated detectable neutralization antibody titers after primary immunization, which was enhanced considerably upon secondary immunization.
Furthermore, there was only a modest innate immune response, at 1 day or 7 days after primary immunization. Surprisingly however, secondary immunization with BNT162b2 induced a much higher magnitude of innate immune response than after primary vaccination 9.
As the earlier study found, and less effective against malignant cells:
This enhanced “secondary innate response,” was characterized by increased levels of IFNγ in the plasma, enhanced transcriptional signatures of innate immunity and antiviral immunity in myeloid cells.
The mechanism:
BNT162b2 stimulates CD8 T cell responses via MDA5-IFNα axis
TLRs are the most studied sensors of pathogen-associated molecular patterns 32. TLR3 and TLR7 recognize double- and single-stranded RNA respectively 33, 34, 35. Therefore, we first examined whether deficiency of these receptors results in a diminished immune response to BNT162b2.
However, immunization of Tlr3−/− or Tlr7−/− mice did not cause a reduction of antibody or T cell responses (Extended Data Fig.5a–c), suggesting that they are not the primary sensors of BNT162b2 in vivo. Previous evidence indicates that the SARS-CoV-2 spike protein binds to and stimulates TLR2 and TLR4 signaling 36, 37.
The mice did not respond via the inflammasome pathway or via cGAS-STING, a cytoplasmic nucleic acid-sensing mechanism. However, MDA5 deficient mice had a much reduced CD8 T cell response, with the concentration of serum total IFNα below their detection limit:
Interestingly, Mda5−/− mice had a striking reduction in the frequency of antigen-specific CD8 T cells as evidenced by fewer class I tetramer specific CD8 T cells and IFNγ-producing CD8+ T cells after primary (Extended Data Fig.9) and secondary immunizations (Fig. 7c, ,d)d) suggesting that the MDA5 sensing of BNT162b2 is responsible for the induction of spike-specific CD8 T cell responses.
Collectively, these data show that MDA5 senses BNT162b2 to induce expression of IFNα, which stimulates antigen-specific CD8 T cell expansion via IFNAR1, to promote high magnitude of antigen-specific CD8 T cells. Thus, MDA5 deficiency leads to IFNα deficiency, which in turn leads to reduced CD8 T cell response after primary vaccination, which then leads to reduced recall responses.
Effects were dose-dependent:
Experiments using a low dose (0.2 μg) of vaccine also resulted in similar B and T cell responses, but the magnitude was 5–10-fold lower than the high dose.
A type 1 IFN response to dose 1 was detected, but suppressed, whereas the MDA5-triggered type 2 IFNγ response from dose 2 was restricted mainly to the T cells rather than ordinary nucleated cells. Is this because T cells are less likely to be transfected and deactivated?
We know you need both type 1 or/and 2 responses to avoid tumor proliferation for different cancer types, and for therapeutics to work optimally:
We extended these findings to show that the CD4 and CD8 T cells were the primary source of serum IFNγ 1-day after secondary immunization, which results in the enhanced myeloid cell activation after the secondary immunization.

This is not the case after transfection with 100% m1Ψ-modified mRNA, which also inhibits pattern recognition of tumor-associated antigens (TAAs):
The source of Type I interferon
Most cells in the body could produce IFN-Is in response to the stimulation of pattern recognition receptors (PRRs) by pathogens. Upon the activation by pathogens, various innate immune cells, including macrophages and DCs [31, 32], can produce IFN-Is. However, non-immune cells such as fibroblasts and epithelial cells also contribute to the production of IFN-Is [33].
PRRs are located on the cell surface, in the cytosol and endosomal compartments (Table 1), and are responsible for recognizing various pathogen-associated molecular patterns (PAMPs), including nucleic acids and non-nucleic acid PAMPs [31].

From: “The crucial regulatory role of type I interferon in inflammatory diseases“ (2023)
https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-023-01188-z
This study from 2001 by Mattei et al. found that you need both type 1 and 2 interferons to mount an optimal anti-tumor response via IL-15 signaling. What’s even more striking is that IL-15 promoted type 2 IFN-y in a paracrine manner even when expressed at almost undetectably low levels.
This implies that a minor “damping” of any immune response may have an unexpectedly disproportionately large suppressive effect on anti-tumor (or viral) responses, even though the signalling proteins involved may not be measurable:
Key takes from “IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation” (2001):
IFN-αβ appeared to regulate the sensitivity of DCs to IL-15, because DC activation in response to IL-15 injection was reduced in IFN-αβR−/− mice. This may be linked to the ability of IFN-αβ to up-regulate IL-15Rα (see below). Alternatively, signals through IFN-αβR and IL-15R could act independently and synergize in DC activation.
The increased expression of IL-15Rα that was observed after IFN-αβ, poly(I:C), or LPS treatment suggests that infection, in addition to augmenting production of IL-15, could enhance the ability of DCs to respond to IL-15.
Small changes in expression of the α-chain, which together with the shared β- and γ-subunits of the IL-2R forms the complete IL-15R, may markedly affect cell sensitivity to IL-15 given its high affinity for IL-15Rα (23).
In fact, the affinity of IL-15Rα for IL-15 is 1000-fold higher than the affinity of IL-2Rα for IL-2. Therefore, enhanced expression of IL-15Rα may allow DCs to respond to very low quantities of IL-15. In this regard, macrophages have been shown to respond to concentrations of IL-15 as low as 10−6 ng/ml (53).
Such a high level of sensitivity to IL-15 may be important, because the difficulties in detecting secreted IL-15 in most studies suggest that IL-15 is indeed released from cells in very small amounts (46, 54).
Furthermore, increased expression of both IL-15 and IL-15Rα in DCs after IFN-αβ or poly(I:C) treatment implies that IL-15 could act in an autocrine or paracrine manner to activate DCs. This is worth considering, as IL-15 can apparently exert autocrine/paracrine effects on macrophages, melanoma cells, and myeloma cells even though secreted IL-15 cannot be detected in the supernatants of these cells (45, 53, 55).
The Th1 response is proinflammatory and, unlike the m1Ψ-associated immune damping Th2 response, can kill tumor cells by activating death receptors on the cell’s surface:
IL-15 treatment stimulated a marked increase in IFN-γ secretion by DCs, acting similarly in this respect to IL-12 (43). This raises the possibility that, in addition to activating DCs, IL-15 treatment may modulate the type of T cell response elicited.
For example, DC-secreted IFN-γ could enhance IL-12 production by macrophages (67), ultimately promoting a Th1-type response.
Another positive feedback loop:
dsRNA, LPS or type 1 IFN-αβ > IL-15 > DCs > type 2 IFN-γ > IL-12 > Th1 proinflammatory response
dsRNA, LPS or type 1 IFN-αβ > IL-15 >DCs > IL-15Rα
PBS is the control:
![FIGURE 7. Enhanced ability of IL-15-treated DCs to stimulate Ag-specific proliferation of naive CD8+ T cells. rmIL-15 or PBS was injected i.v. into WT 129 (A and C) or IFN-αβR−/− mice (B and D), and splenic DCs were isolated 4 h later. Responder CD8+ T cells were purified from lymph nodes of 2C mice and cultured together with either a constant number of DCs (104) and different concentrations of specific peptide (A and B) or a constant concentration of peptide (0.05 nM) and different numbers of DCs (C and D). After 4 days of culture, [3H]thymidine was added, and the cells were cultured for an additional 16 h before harvesting. Each data point represents the amount of [3H]thymidine uptake of a triplicate culture ± SD. ○, Splenic DC from PBS-treated mice; ▪, splenic DC from rmIL-15-treated mice.](https://substackcdn.com/image/fetch/$s_!xKnN!,f_auto,q_auto:good,fl_progressive:steep/https%3A%2F%2Fsubstack-post-media.s3.amazonaws.com%2Fpublic%2Fimages%2Fe4ecb606-3215-4a17-bc69-e021b14fb338_520x675.jpeg "FIGURE 7. Enhanced ability of IL-15-treated DCs to stimulate Ag-specific proliferation of naive CD8+ T cells. rmIL-15 or PBS was injected i.v. into WT 129 (A and C) or IFN-αβR−/− mice (B and D), and splenic DCs were isolated 4 h later. Responder CD8+ T cells were purified from lymph nodes of 2C mice and cultured together with either a constant number of DCs (104) and different concentrations of specific peptide (A and B) or a constant concentration of peptide (0.05 nM) and different numbers of DCs (C and D). After 4 days of culture, [3H]thymidine was added, and the cells were cultured for an additional 16 h before harvesting. Each data point represents the amount of [3H]thymidine uptake of a triplicate culture ± SD. ○, Splenic DC from PBS-treated mice; ▪, splenic DC from rmIL-15-treated mice.")
The BNT162b2-MDA5 enigma
We saw from the earlier study “that the MDA5 sensing of BNT162b2 is responsible for the induction of spike-specific CD8 T cell responses”. This is an enigma because according to multiple studies even MDA5 activation should also be “dampened” by 100% m1Ψ.
Key takes from “Innate immune mechanisms of mRNA vaccines” (2022) by Verbeke et al.:
Modified nucleosides can also dampen activation of various dsRNA sensors, including TLR3, retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), protein kinase R (PKR), and 2’-5’-oligoadenylate synthetase (OAS) (Anderson et al., 2010, 2011; Karikó et al., 2005; Mu et al., 2018).
Recognition of uridine-containing RNA is associated with increased expression of pro-inflammatory cytokines, particularly type I IFN (Karikó et al., 2005, 2011), which promotes the expression of RNA sensors even further and can lead to inhibition of antigen expression from the mRNA via the activities of PKR and OAS.
A plausible explanation is straight from the pharma clownworld that regulatory capture leads to: contamination with double-stranded RNA.

Another mRNA vaccine component that can significantly contribute to deleterious innate immune recognition is dsRNA. In vitro transcription (IVT) by T7 RNA polymerase tends to yield not only the desired mRNA, but also a variety of unwanted RNA species, including various short abortive transcripts as well as antisense RNAs transcribed from the promoter-less end of the DNA template (Karikó et al., 2011; Mu et al., 2018). This can lead to the formation of dsRNA, which induces a potent inflammatory response and translational blockade via the recognition of various intracellular receptors (OAS, PKR, RIG-I, MDA5, TLR3, DHX33, etc.) (Anderson et al., 2010, 2011; Durbin et al., 2016; Karikó et al., 2011; Mitoma et al., 2013).
Secondary structures also carry their own risk of promoting cancer, and we also discussed these in our review.
Double-stranded secondary and tertiary structures could also form based on the sequence of the mRNA.
… The importance of RNA structural elements in shaping the alternatively spliced transcriptome in tumorigenesis was highlighted recently (Zhang et al., 2021). Moreover, the prevalence of G4 structures within the human transcriptome hints at their significant involvement in splicing regulation suggesting a major modulatory role of rG4s in cancer. Single nucleotide polymorphisms (SNPs) within G4 sequences can disrupt their secondary structure leading to altered splicing events often in cancer-related genes and in turn to tumorigenesis or tumor progression and metastasis (Beaudoin and Perreault, 2010).
… an SNP polymorphism within the G4 sequence modifies the binding capacity of ligands resulting in the retention of intron 2 and the expression of elongated TP53 variants that dominate over the fully spliced isoform. Increased expression of such variants leads to impairment of TP53 functions in suppression of cell growth and thus controlling tumor formation and progression (Perriaud et al., 2014).
From: “Aberrant splicing in human cancer: An RNA structural code point of view“ (2023)
They could have reduced it down to a safe level. Even Karikó et al warned of this:
First, as mentioned above, the inclusion of modified nucleosides can reduce the activation of PKR and OAS (Anderson et al., 2010, 2011). Second, dsRNA can be removed from IVT mRNA by certain purification methods. While dsRNA is not removed by common alcohol precipitation methods, it can be significantly reduced by high performance liquid chromatography (HPLC) (Karikó et al., 2011) or simpler methods such as adsorption on cellulose (Baiersdorfer et al., 2019) or RNase III digestion (Foster et al., 2019).
For the safer 0% m1Ψ unmodified mRNA you don’t have much choice. Whereas from pharma’s viewpoint, with the m1Ψ-modified product, dsRNA contamination is less of a problem as the m1Ψ itself may help reduce it, as well as a side effect being the stimulation of CD8+ T cell responses:
It appears critical to reduce dsRNA from unmodified IVT mRNA to achieve a high level of protein production in vitro in DCs or in vivo in mice, but it may be less critical for m1Ψ-modified IVT mRNA (Baiersdorfer et al., 2019; Karikó et al., 2011; Nelson et al., 2020).
Incorporation of m1Ψ and elimination of dsRNA can both dampen type I IFN responses upon intravenous administration of mRNA-iLNP products into mice (Nelson et al., 2020).
Interestingly, the incorporation of certain modified nucleosides such as Ψ, m1Ψ, and 5-methylcytidine can also reduce the formation of dsRNA byproducts in T7-transcribed mRNA preparations (Mu et al., 2018).
“You can get it if you really want, but you must try”:
Lowering the Mg2+ concentration (from 30 mM to 5 mM) in the IVT reaction mixture and performing the IVT at higher temperature (∼50–55°C vs. 37°C) with a thermostable T7 RNA polymerase also reduces the dsRNA contaminants in the IVT reaction (Mu et al., 2018; Wu et al., 2020), and it is theoretically possible that the T7 enzyme could be mutated or replaced with an RNA polymerase that produces less dsRNA, or none at all, thus facilitating the economical manufacture of high-quality, good manufacturing practice (GMP)-grade mRNA.
Meulewaeter et al. also discussed this:
... As an outstanding example, a study on the mechanisms of BNT162b2 (mRNA COVID vaccine developed by Pfizer/BioNTech) revealed that the melanoma differentiation-associated protein 5 (MDA5)–IFN-I signaling pathway was crucial for the vaccine’s capacity to elicit CD8+ T cells.70 As it has been described that long-dsRNA is sensed by MDA5,71 it is likely that dsRNA contaminants that might be present in the BNT162b2 vaccine might trigger the MDA5-IFN pathway, but this has yet to be proven. These findings suggest that a mere modification of the uridine nucleotide may not be sufficient to entirely eliminate the inflammatory nature of mRNA.
From “Considerations on the Design of Lipid-based mRNA Vaccines Against Cancer“ (2024)
https://www.sciencedirect.com/science/article/pii/S0022283623004965
The regulators were aware of the risk. But as Fizer performed the Process 1 to Process 2 “switcheroo” they were hoodwinked, and of course, there was never any follow-up or product recall:
Specification
The active substance specifications contain tests for appearance (clarity, coloration (Ph. Eur.)), pH (Ph. Eur.), content (RNA Concentration) (UV Spectroscopy), Identity of Encoded RNA Sequence (RT-PCR), RNA Integrity (Capillary Gel Electrophoresis), 5’- Cap (RP-HPLC), Poly(A) Tail (ddPCR), Residual DNA Template (qPCR), dsRNA (Immunoblot), Bacterial Endotoxin (Ph. Eur.) and Bioburden (Ph. Eur.). The proposed specification for active substance is considered acceptable for authorisation with respect to the attributes chosen for routine release testing. During the procedure the specification limits for a number of attributes were tightened in response to questions.
… The rationale used to establish the acceptance criteria is described in detail and based on a limited data set representative of BNT162b2 active substance manufactured at the intended commercial scale and process. From the available data, mRNA integrity, dsRNA and Poly(A) tail acceptance criteria are considered in relation with batches used in clinical studies and with the demonstrated manufacturing capability and need to be re-assessed and revised as appropriate as further data becomes available (SO2).
… 11. The MAH should provide a summary of the validation/verification status of the immunoblot analytical procedure used to detect double stranded RNA (dsRNA) in BNT162b2 active substance.
From: “EUROPEAN MEDICINES AGENCY. Assessment report Comirnaty Common name: COVID-19 mRNA vaccine (nucleoside-modified)“ (19th February 2021)
It gets worse. A periodic benefit-risk evaluation report (PBRER) for the now-withdrawn Astrazeneca COVID-19 “vaccine” from 2022 links dsRNA contamination to the risk of myocarditis.
Of course, every case that was seen in relation to the AZ product was “unrelated”, because there was no other possible mechanism for “circulating spike protein” to be cardiotoxic:
Mechanism of Action of Myocarditis and COVID-19 VACCINE ASTRAZENECA Myocarditis is an inflammatory diseases of the heart muscles. The possible mechanism by which COVID-19 VACCINE ASTRAZENECA may cause myocarditis could be related to inflammation, autoimmunity or a combination of both. Co-morbidities may be a contributing factor which could feed into an inflammatory mechanism as pericytes expressing high levels of the ACE2 receptor and levels have been found to be increased in diseases Chen et al 2020 In animal studies with COVID-19 VACCINE ASTRAZEENCA, circulating spike protein was measured and can be detected after vaccination. Inflammation through activation of the innate pathway through dsRNA has also been implicated for mRNA vaccines Milano et al 2021. Whilst dsRNA is not applicable to COVID-19 VACCINE ASTRAZENECA, adenovirus vectors do activate innate pathways which could be another possible explanation for COVID-19 VACCINE ASTRAZENECA Zhu et al 2007, Muruve 2004.
Based on the cumulative review of safety report cases, AstraZeneca opinion is that the MOA for myocarditis is unrelated to COVID-19 VACCINE ASTRAZENECA.
From: “COVID-19 VACCINE ASTRAZENECA (AZD1222) VAXZEVRIA™ Periodic Benefit-Risk Evaluation Report“ (2022)
dsRNA contamination and myocarditis
By as early as December 2021, a year after the mass rollout, Milano et al. (above) had researched enough studies to write a review for “Future Virology” on possible mechanisms. And this is without even considering the effects of plasmid DNA/SV40 promoter or lipopolysaccharide endotoxin contamination.
Key takes from “Myocarditis and COVID-19 mRNA vaccines: a mechanistic hypothesis involving dsRNA“ (2021):
Abstract
While tolerance to COVID-19 vaccination is considered satisfactory, a phenomenon of myocarditis, although rare, is becoming a safety concern in mRNA COVID-19 vaccination. The presence of low residual levels of double-strand RNA (dsRNA) has been reported in mRNA COVID-19 vaccine preparations. dsRNA is a known inducer of immune-inflammatory reactions. dsRNA present in vaccine nanoparticles may be suspected to be at the origin of the still unexplained cases of myocarditis.
While the simplicity of the approach of synthesizing in vitro-transcribed (IVT) mRNA is appealing, technical difficulties remain, including impurities and particularly the generation of dsRNA contaminants [16] as indicated above.
The current methods used to purify IVT mRNA vaccine preparations vary in terms of technical performance and, at best, allow the removal of 90% of dsRNA when using HPLC, as reported by the developers of mRNA vaccines [17]. According to reports and vaccine developers, the presence of short segments of dsRNA at low level along with purified mRNA cannot be totally ruled out [17].
Notably, dsRNA is known to be a strong exogenous inducer of immune-inflammatory reactions involving well-identified intracellular signaling cascades and mediators (Figure 1). dsRNA is detected by antigen-presenting cells, endothelial cells and the airway epithelium [18], and gives rise to dose-related innate immune activation [17].
There was no informed consent with respect to unavoidably high levels of dsRNA contamination and its known risks:
The presence of dsRNA, even at low levels, raises questions about its possible favorable and unfavorable consequences, bearing in mind that a new, improved technique to remove dsRNA has been reported recently [16].
This improved mRNA purification method, hardly transposable on an industrial scale, consists in the adsorption of dsRNA contaminants to cellulose and could work better than chromatographic methods of removing dsRNA from IVT mRNA samples.
Regarding the consequences of the presence of dsRNA (Figure 1), on the one hand, dsRNA could provide an advantage (intrinsic adjuvant) by inducing a high immunological response, which is obtained with COVID-19 mRNA vaccines.
However, this remains an hypothesis and there are no dedicated studies evaluating a possible benefit of dsRNA in mRNA vaccines conferring an added protection against SARS-CoV-2.
On the other hand, dsRNA could boost the induction of some uncontrolled and potentially detrimental immune-inflammatory reactions, such as myocarditis (Figure 1).
The LNP/synthetic mRNA platform has been found to increase the risk of myocarditis through multiple mechanisms. dsRNA contamination probably adds to the list, even at low levels:
This leads to the next unavoidable question: can the presence of dsRNA in mRNA vaccines, even at low concentration, explain some of the undesired effects?
In the case of myocarditis, it should be noted that when packaged in lipid nanoparticles, dsRNA is preferentially transferred to phagocytic monocytic-derived cells, such as macrophages and dendritic cells, which are key actors in immunity [24].
Recent studies indicate that precursors of dendritic cells patrol the blood and communicate with immature dendritic cells residing in peripheral tissues such as the kidneys, skin and myocardium. Dendritic cells trigger immune responses in lymphoid tissues upon early sensing of infectious pathogens.
Globally, dendritic cells form a sentinel network that modulates immune responses with the distinct ability to produce protective immunity or tolerance to self. Dendritic cells also play a major role in the pathophysiology of inflammatory diseases. As a result, the uptake of dsRNA by dendritic cells is suspected of triggering immune reactivity and inflammatory reactions.
The oncologists must have missed this nugget at the time:
The proinflammatory role of dsRNA, even at low concentration in mRNA COVID-19 vaccines, can constitute a true risk for myocarditis. This risk should thus be considered when prescribing vaccination in cancer patients who have been treated or are under an anticancer treatment that threatens the heart.
MDA5 and autoimmunity
MDA5 was first described in 2002. Its interactions are far more complex than I’ve described here so far, and even now all the mechanisms aren’t fully understood.
In 2019, Junior et al. reviewed how MDA5 has to balance out immunity against viral infections with autoimmunity. If you carry a mutation in the gene encoding MDA5 then you may be at an elevated risk of autoimmune disease or death on vaccination.
The full paper does a deep dive into why some RNA activates MDA5, but not others.
Key takes from “A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation“ (2019):
Recent work showed that mutations in the IFIH1 gene, encoding MDA5, lead to interferon-driven autoinflammatory diseases. Together with observations made in cancer cells, this suggests that MDA5 detects cellular RNAs in addition to viral RNAs.
It is therefore important to understand the properties of the RNAs which activate MDA5. New data indicate that RNA length and secondary structure are features sensed by MDA5. We review these developments and discuss how MDA5 strikes a balance between antiviral immunity and neuroinflammation.
This is probably describing what viruses you are at greater risk of contracting after BNT162b2, the RIG-I>IFN type 1 triggers:
Although RIG-I and MDA5 share similar domains, they detect different viral infections [18]. For example, RIG-I detects infection with orthomyxoviruses, such as influenza A virus, while MDA5 senses picornavirus infection.
In contrast to RIG-I, the mechanisms that allow MDA5 to recognise viral RNAs while avoiding cellular RNAs are less well understood. Biochemical and structural work using recombinant MDA5 protein demonstrated that MDA5 senses RNA length and secondary structure 19, 20, 21.
If cellular RNA’s trigger MDA5 then you are at risk of various autoimmune diseases:
Understanding molecular features of MDA5-stimulatory RNAs is important for another reason: in addition to its protective role in antiviral defence, MDA5 has been implicated in autoimmune and autoinflammatory diseases such as type 1 diabetes (T1D), systemic lupus erythematosus (SLE) and Aicardi–Goutières syndrome (AGS) 2, 3, 22.
Moreover, new data indicate that MDA5 is activated during some forms of cancer treatment 23, 24 or in settings where mitochondrial RNA degradation is compromised [17]. These observations highlight that, in some circumstances, cellular RNAs trigger MDA5.
CLIP analysis: cross-linking and immunoprecipitation, which is used to identify the sites bound by a specific RNA-binding protein on endogenous RNAs:
Although CLIP analysis reveals both activating and nonactivating RNA ligands, it may be particularly informative as it allows interrogation of MDA5-associated RNAs in virus infected cells.
One of the hypotheses for why 100% m1Ψ-modified mRNA helps to promote cancer is by binding to, but not triggering RLRs such as RIG-I, whilst at the same time it is competing with non-vax antigens. These could be viral RNAs, viral RNA-binding proteins, or tumor-associated. You could think of the m1Ψ-modified mRNA as mimicking one of the immune suppressive actions of the virus.
Very few papers have so far mentioned competitive inhibition. Substitute 100% m1Ψ-modified mRNA for the viral RNA-binding proteins:

This is important because viral RNAs are typically coated by viral proteins, and RLRs have been suggested to compete with viral RNA-binding proteins [54].
As such, the actual RNA agonist recognised by MDA5 in infected cells is likely to be determined by a combination of factors, including (i) the RNA binding preferences of MDA5, (ii) accessory host factors such as LGP2 [53], DHX29 [55], and LncITPRIP-1 [56] that facilitate RNA binding by MDA5, (iii) the abundance and properties of viral RNA-binding proteins [54], and (iv) viral evasion strategies such as RNA methylation [57] and RNA degradation [58].
For vulnerable groups, dsRNA contamination is likely to exacerbate any of the following conditions. T1D is autoimmune type 1 diabetes; SLE is systemic lupus erythematosus:
In addition to its role in the detection of viral infections, MDA5 has also been implicated in a number of sterile, inflammatory conditions. For example, single-nucleotide polymorphisms (SNPs) in IFIH1, the gene encoding MDA5, have been associated with T1D, psoriasis, rheumatoid arthritis, vitiligo, multiple sclerosis, and SLE 62, 63, 64, 65, 66, 67, 68. How IFIH1 risk alleles relate to the development of these diseases is not fully understood but it is likely that this involves chronic induction of type I IFNs, which then initiate or enhance autoinflammation and autoimmune responses.
Concluding Remarks
MDA5 is an important RNA sensor implicated in the detection of viral infections, in a range of autoimmune and autoinflammatory diseases, as well as in cancer. Recent efforts in identifying an RNA molecular pattern detected by MDA5 suggest that length and secondary structure of RNA are important determinants; however, we speculate here that activation of MDA5 in some settings may require more than simply dsRNA and that a yet-to-be identified consensus RNA PAMP detected by MDA5 may exist.
Dr Sabine Stebel just posted a deep dive into vax-induced skin diseases, many of which are autoimmune in nature and linked to dysregulation of IFNs:
TLR7/9 versus TLR3/MDA5 signaling
I was looking for a paper that compares receptor signaling and tumor cell proliferation rates. This paper doesn’t quite do that, but it does indirectly by discussing how TLR7/9 signaling promotes dendritic cell activation. These do have a role in cancer immunity.
Key takes from “TLR7/9 versus TLR3/MDA5 signaling during virus infections and diabetes“ (2011) by Swiecki et al.:
pDCs are thought to have a critical role in antiviral defense because of their ability to rapidly secrete large amounts of IFN-I through TLR7/9 signaling.
dsDNA contamination may trigger the MDA5 signaling pathway, but it is also linked to autoimmune diseases:
In the absence of infection, IFN-I production by pDCs or from signaling through dsRNA sensors has been implicated in the pathogenesis of autoimmune diseases such as diabetes.
A recent study demonstrates that although pDCs are a source of IFN-I in vivo, their overall contribution to viral containment is limited and time-dependent, such that additional cellular sources of IFN-I are required to fully control viral infections. dsRNA sensors, such as TLR3 and MDA5, provide another important trigger for antiviral IFN-I responses, which can be exploited to enhance immune responses to vaccines.
What viruses are you at greater risk of due to m1ψ inhibition of TLR7>IFN1 signaling?
Murine cytomegalovirus (MCMV); Lymphocytic choriomeningitis (LCM); Herpes simplex virus (HSV); Indiana vesiculovirus, formerly Vesicular stomatitis Indiana virus (VSIV or VSV); human immunodeficiency virus (HIV); Human T-lymphotropic virus type 1 (HTLV-1); Mouse Hepatitis Virus (MHV); hepatitis C virus (HCV); Newcastle disease virus (NDV); influenza, Sendai virus (SeV); coxsackievirus (CVB1-CVB6); pneumovirus; Epstein–Barr virus (EBV):
Many viruses can promote pDC activation and secretion of IFN-I via the TLR7/9-MyD88-IRF7 pathway, such as MCMV, LCMV, HSV, VSV, HIV, HTLV-1, MHV, HCV, NDV, influenza, SeV, CVB, pneumovirus, and EBV [43–62].
Although TLR7/9 signaling promotes pDC activation and IFN-I secretion, TLR7 and TLR9 are also expressed in other immune cells, including monocytes, classical DCs, and B cell subsets [63, 64]. Thus, it is important to clarify to what extent pDCs contribute to innate and adaptive antiviral responses elicited by viruses sensed by the TLR7/9 pathway.
“Early” is essential for efficiently supressing viral infections:
In a recent study, BDCA2-DTR Tg mice were challenged with representative RNA and DNA viruses, VSV and MCMV, respectively [79]. It was shown that pDCs provide an early but limited source of IFN-I during VSV and MCMV infections (Fig. 1). In the case of VSV infection, IFN-I produced by pDCs limits viral replication and perpetuates the survival and accumulation of antigen-specific CD8+ T cells.
RIG-I has been shown to detect paramyxoviruses (SeV, NDV, respiratory syncytial virus, and Measles virus); orthomyxovirus (influenza A and B viruses); rhabdovirus (VSV and Rabies virus); flavivirus (Japanese encephalitis virus, HCV, WNV, and DV); filovirus (Ebola virus); reovirus; and metapneumovirus [106, 123, 128–144]. Recent studies have shown that RIG-I also recognizes DNA viruses by detecting RNA intermediates generated through the RNA polymerase III-mediated transcription of dsDNA [145, 146]. MDA5 detects picornaviruses such as EMCV, CVB, Mengo virus, and Theiler virus, as well as murine norovirus 1 [132, 146–149]. MDA5 is also involved in the recognition of WNV, DV, reovirus, SeV, MHV, Measles virus, and LCMV [106, 134, 138, 139, 150–153].
In Part I I reviewed a study showing how transfection knocks out this response:
TLR3 contributed to NK cell activation by promoting IFN-I and IL-12 release by hematopoietic cells. MDA5 acted indirectly by promoting stromal cell release of IFN-I, which activated NK cells. Thus, in mice, poly(I:C) stimulates NK cell responses by inducing the production of various cytokines in a manner dependent on the cell type and dsRNA sensor.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3177694/
Parting shots
Ψ‐linked diseases - using the wrong codon
Xia wrote a critical evaluation of the technology: “Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines” (2021)
Hat tip to DR AH KAHN SYED (pseudonym) for the link in “How many coincidences...” .
Key takes:
The direct causative pathogenic association between decreased Ψ levels and human disorders, particularly neuronal dysfunctions and resulting behavior defects, has been confirmed [26] (Fig. 2).
The list of Ψ‐linked diseases includes mitochondrial myopathy and sideroblastic anemia caused by mutations in the PUS1 gene that impairs pseudouridylation of specific tRNAs and PUS1‐dependent mRNA expression [27, 28, 29].
Autosomal recessive mental retardation (MRT55) is related to PUS3 mutations while intellectual disorders are associated with missense mutations in PUS7 [26, 30].
Together with other factors, the PUS10 gene was identified as a risk locus in Crohn's disease, an inflammatory disease [31].
Recently, clinical‐relevant mutations in DKC1 and NOP10 have been proven to reduce pseudouridylation in rRNAs and cause nephrotic syndromes in fish [24].
In addition, aberrant pseudouridylation profiles have been linked to cancer formation [32].
There are two lines of evidence suggesting that CGG is not the optimal codon. The first involves the codon usage of human ribosomal protein genes (“RP” in Table 1) which are known to be highly expressed. These genes prefer CGC codons (Table 1).
The second and more direct evidence is from codon usage of genes highly expressed in skeletal muscle cells (which are relevant here because the vaccine mRNA is injected and carried by the lipid nanoparticles into skeletal muscle cells to be translated, although vaccine mRNA could also be carried to some other tissues).
Therefore, the CGC codon preferred by ribosomal protein genes are also preferred by highly expressed muscle genes. Other protein-coding genes that are highly expressed are the two isoforms of human elongation factor 1α (hEF1A1 and hEF1A2), and poly(A)-binding protein (hPABPC1).
They also use more CGC than CGG (CGC:CGG are 3:0 for hEF1A1, 8:6 for hEF1A2, and 14:4 for hPABPC1). These multiple lines of evidence suggest that CGC is a better codon than CGG.
The designers of the mRNA vaccines (especially mRNA-1273, Table 1) chose a wrong codon as the optimal codon.
mRNA cancer vaccines
This story was all over the MSM today:

But first, a reality check:
No miracles
While mRNA coronavirus vaccines may have surpassed expectations, cancer experts caution against getting too optimistic over mRNA’s application in cancer.
Zaks sees a threefold challenge: “First, can an mRNA vaccine generate a T-cell response? Second, are we generating the response against the right antigen? And three, is it good enough?”
The reality is that while existing immunotherapies extend some people’s lives, they aren’t effective for everyone.
“We don't even understand fully why all the various different immunotherapies that we've got don't work for some people and do work for other people,” said Godfrey at CRUK. “I'm sure [mRNA cancer vaccines] will be successful in the future for a subset of people. But it is by no means a miracle cure.”
In the U.S. and the EU, there is just a handful of non-mRNA vaccines approved to treat cancers — the BCG vaccine against tuberculosis is used to treat bladder cancer; Amgen’s Imlygic is given to patients with advanced melanoma; and Provenge has been used to treat prostate cancer. However, Provenge’s marketing authorization was withdrawn in the EU in 2015 for commercial reasons.
Phase 3 trials of non-mRNA cancer jabs from Germany’s Merck KGaA and Britain's GSK have meanwhile failed to improve patient survival.
Experts believe mRNA cancer vaccines will run up against the challenges faced by other immunotherapies. “It’s a complex area. I believe that we will certainly have some positive data with mRNA approaches but it’s not simple,” said Matti Aapro, immediate past president of the European Cancer Organization (ECO). He said it’s likely mRNA vaccines would be complementary to more traditional immunotherapy.
One problem researchers face is identifying which antigens the vaccine should target, and here better bioinformatics tools are needed, said Fabrice André, an oncologist at Gustave Roussy Cancer Center in France.
… The eventual jabs are going to be pricey, and difficult value-for-money questions are sure to arise. For existing immunotherapies, the costs can be eye-watering: with the bill for CAR-T cell therapy can run to $1 million while checkpoint inhibitors often cost over $100,000 a year. The price of any future mRNA cancer vaccine is unknown but it’s certainly not going to be cheap.
“If something's costing a million pounds per treatment for a personalized vaccine, and its effectiveness isn't much better than standard chemotherapy, which doesn't cost very much, you've got that decision about, actually, is this worth investing in?” said CRUK’s Godfrey.
Efforts are underway to address this. For example, CRUK is working with the U.K.’s National Health Service on developing potential new models where payments are linked to drug performance. These envision a sliding scale — if a drug starts to do really well they can charge more; if not, costs come down.
… “If you want to charge for something, you've got to show that it brings value," said Zaks. "And I think it's good that we have to prove that the vaccines work and that they actually bring value.”
From: “After coronavirus vaccine breakthrough, mRNA pioneers pivot to cancer“ (2022)
https://www.politico.eu/article/mrna-cancer-vaccine-coronavirus/
By “pricey” Dr. Makis puts this currently at about $500,000:

Yao, Xie & Xia recently published a review of the clinical trials and developments in the field of mRNA cancer vaccines: “Recent progress in mRNA cancer vaccines“ (2024).
Key takes:
Collectively, mRNA vaccines are emerging as a central focus in cancer immunotherapy, offering the potential to address multiple challenges in cancer treatment, either as standalone therapies or in combination with current cancer treatments.
Case reports and analysis do not agree with these statements, due to plasmid DNA + SV40 promoter contamination and evidence of persistent expression:
One of the key advantages of mRNA vaccines is their exceptional safety profile. Unlike DNA-based therapies, mRNA vaccines do not integrate into the host genome, thereby negating the risk of insertional mutagenesis.
Get this wrong, which is easy, and you risk off-target autoimmune diseases and aggressive recurrences due to immune escape:
Furthermore, mRNA’s ability to concurrently encode multiple antigens boosts vaccine efficacy and paves the way for the development of personalized vaccines.
Reverse transcription and integration?
The transient nature of mRNA expression ensures that antigen stimulation is temporary, effectively reducing the likelihood of chronic inflammations or autoimmune responses.3
Even for cancer patients “rapid design and synthesis” means a lack of long term clinical safety and efficacy data, when compared to existing therapeutics:
Additionally, the rapid design and synthesis of mRNA vaccines enable swift responses to evolving oncological challenges, including customizing vaccines to the unique tumor characteristics of individual patients, marking a significant stride toward personalized cancer therapy.4
Although mRNA cancer vaccines are showing considerable promise, their broader application is hindered by several challenges. Challenges among these are the inherent instability of mRNA, the need for efficient and targeted delivery systems, and the possibility of immune tolerance, as well as evasion by tumor cells. These challenges present significant obstacles that are the focus of current research efforts.5
… early mRNA molecules faced limitations due to instability and low expression efficiency. Unmodified mRNA was initially noted for its high immunogenicity, activating innate immune responses through toll-like receptors (TLR3, TLR7, TLR8, and TLR9).14–17
The irony and perfidies of these statements:
The subsequent incorporation of modified nucleosides (such as m5C, m6A, m5U, s2U, and pseudouridine) into mRNA formulations markedly reduced their immunogenicity, with the complete replacement of uridine with pseudouridine improving mRNA stability and translation efficiency.18,19
The scientists who made these breakthrough findings, Drs. Katalin Karikó and Drew Weissman, were awarded the 2023 Nobel Prize in Physiology or Medicine for their discoveries concerning nucleoside base modifications that enabled the development of effective mRNA vaccines against COVID-19.
Pembrolizumab (Keytruda) is a PD-1 immune checkpoint inhibitor:
The progression of mRNA-4157 in combination with pembrolizumab to Phase III clinical trials for melanoma patients represents a significant milestone in the field of personalized mRNA cancer vaccines.
Lets see how the trials went, although this sort of report is often goal-seeked from the desired “result” by cherry picking, using bridging studies, using multiple studies to “bury” AE’s, and by tweaking the assessment criteria used to measure “success”.
The side effects reported from the various KEYNOTE Keytruda trials could fill a book, these are just a few:

Serious adverse reactions occurred in up to 50% of patients in KEYNOTE-869, and discontinuation in up to 32% of patients. The authors acknowledged that side effects (which can including death) remained, and the mRNA product added to the list of AE’s with its own, 25% vs 20%, or a 25% increase.
60% of patients reported fatigue, and grade 3 “fatigue” is quite severe:
Symptomatic, capable of only limited self care, confined to bed for more than 50% of waking hours or completely bed or chair bound.
Disabling fatigue or bedridden.
Fatigue is quite alarming in itself. With SARS-CoV-2 mRNA vaccination, it is a symptom of cancer-associated vaccine poisoning (post-acute COVID-19 vaccination syndrome, or PACVS) and severe metabolic dysfunction (ie. damaged mitochondria).
Blood markers in one study of PACVS showed increased levels of oncogenic IL-6, increased Angiotensin II type 1 receptor antibodies, and decreased alpha-2B adrenergic receptor antibodies.
Note: As with today’s big story about a colon cancer vaccine trial, “Complete resection” means that the mRNA-4157 + Keytruda treatment isn’t being assessed for efficacy at treating cancer, but how effective it is at preventing recurrence within three years (a short period to conclude anything) after surgical removal.
Certainly no miracle cure, and you still face years of awful side effects after surgery:
Moderna And Merck Announce mRNA-4157 (V940) In Combination with Keytruda(R) (Pembrolizumab) Demonstrated Continued Improvement in Recurrence-Free Survival and Distant Metastasis-Free Survival in Patients with High-Risk Stage III/IV Melanoma Following Complete Resection Versus Keytruda at Three Years
December 14, 2023
At a median planned follow-up of approximately three years, mRNA-4157 (V940) in combination with KEYTRUDA reduced the risk of recurrence or death by 49% (HR=0.510 [95% CI, 0.288-0.906]; one-sided nominal p=0.0095) and the risk of distant metastasis or death by 62% (HR=0.384 [95% CI, 0.172-0.858]; one-sided nominal p= 0.0077) compared to KEYTRUDA alone in stage III/IV melanoma patients with high risk of recurrence following complete resection.
… Adverse events observed with mRNA-4157 (V940) in KEYNOTE-942 remain consistent with those previously reported. At a median planned follow-up of approximately three years, the number of patients reporting treatment-related Grade ≥ 3 adverse events were similar between the arms (25% for mRNA-4157 (V940) in combination with KEYTRUDA vs 20% for KEYTRUDA alone). The most common adverse events of any grade attributed to mRNA-4157 (V940) were fatigue (60.6%), injection site pain (56.7%), and chills (49%).
… mRNA-4157 (V940) is a novel investigational messenger RNA (mRNA)-based individualized neoantigen therapy (INT) consisting of a synthetic mRNA coding for up to 34 neoantigens that is designed and produced based on the unique mutational signature of the DNA sequence of the patient's tumor. Upon administration into the body, the algorithmically derived and RNA-encoded neoantigen sequences are endogenously translated and undergo natural cellular antigen processing and presentation, a key step in adaptive immunity.
… In KEYNOTE-869, when KEYTRUDA was administered in combination with enfortumab vedotin to patients with locally advanced or mUC and who are not eligible for cisplatin-based chemotherapy (n=121), fatal adverse reactions occurred in 5% of patients, including sepsis (1.6%), bullous dermatitis (0.8%), myasthenia gravis (0.8%), and pneumonitis (0.8%). Serious adverse reactions occurred in 50% of patients receiving KEYTRUDA in combination with enfortumab vedotin; the serious adverse reactions in ≥2% of patients were acute kidney injury (7%), urinary tract infection (7%), urosepsis (5%), hematuria (3.3%), pneumonia (3.3%), pneumonitis (3.3%), sepsis (3.3%), anemia (2.5%), diarrhea (2.5%), hypotension (2.5%), myasthenia gravis (2.5%), myositis (2.5%), and urinary retention (2.5%). Permanent discontinuation of KEYTRUDA occurred in 32% of patients. The most common adverse reactions (≥2%) resulting in permanent discontinuation of KEYTRUDA were pneumonitis (5%), peripheral neuropathy (5%), rash (3.3%), and myasthenia gravis (2.5%). The most common adverse reactions (≥20%) occurring in patients treated with KEYTRUDA in combination with enfortumab vedotin were rash (71%), peripheral neuropathy (65%), fatigue (60%), alopecia (52%), weight loss (48%), diarrhea (45%), pruritus (40%), decreased appetite (38%), nausea (36%), dysgeusia (35%), urinary tract infection (30%), constipation (27%), peripheral edema (26%), dry eye (25%), dizziness (23%), arthralgia (23%), and dry skin (21%).
Returning to key takes from “Recent progress in mRNA cancer vaccines”.
There has been a lot of focus on increasing lifespan and stability, whilst decreasing immunogenicity:
Integrating T7 RNA polymerase with VCE simplifies cap-modified mRNA synthesis, reducing the need for expensive analogs.28 Continuous cap analog optimization has significantly improved mRNA stability and transcription, enhancing its research and application potential (Table 2).29–31
Furthermore, a recent study shows a shift from cap1 to cap2 in mRNA caps with age, decreasing RIG-I receptor activation and increasing mRNA stability.32 Incorporating cap2 may reduce immunogenicity and extend mRNA lifespan.
The Poly(A) tail, characterized by a long repetitive polyadenylate sequence, plays a crucial role in defending mRNA against enzymatic deacetylation and degradation, thereby augmenting its stability and translational efficacy.31
The incorporation of short sequences or thiophosphate between polyadenylate sequences has been shown to further enhance stability. For example, the Poly(A) tail sequence of BNT162b2 comprises a structure of 30A + 10GCAUAUGACU +70A.49,50
Additionally, a recent study demonstrated that a Poly(A) tail containing 20% cytidine can protect mRNA from deacetylase activity and significantly improve the translation efficiency and half-life of mRNA.51,52
As we saw in part I, you cannot standardise protocols because you can either design for protein overexpression OR for immune cell recruitment, but not both, as these are mutually exclusive.
I need to ask why the obsession with protein production? The need for combination therapies shows that cancer antigen presentation is probably not the limiting factor in recruiting immune cells and for turning cold tumors hot.
You may consider using cancer mRNA vaccines to treat your patient without other therapies, but how do you know it will work, that the tumor being presented is susceptible? The answer is that you don’t.
And neoantigens are a product of random mutations in tumor DNA. By their very nature, no cancer vaccine can be universal enough to anticipate these without risking off-target autoimmune diseases, which could easily prove fatal (eg cardiovascular, pulmonary, liver or kidney involvement) or debilitating (neurological).
Optimization strategies for mRNA structure can be synergistically employed to enhance protein production. The advancement of mRNA vaccines underscores the need for a universally standardized mRNA design and screening protocol.
Comprehensive screening and precise customization of target-specific sequences hold the promise of diverse enhancements in future mRNA designs. This approach enables personalized adaptations to specific cell types and unique microenvironment, thereby maximizing the protein synthesis efficiency of each mRNA transcript.
No off-switch. What could possibly go wrong?
Besides linear mRNA, several other types of RNA vectors have advanced quickly (Figure 1). One such promising vector is the self-amplifying RNA (saRNA), which is derived from a virus and carries the RNA polymerase sequence. This saRNA exhibits self-amplification and translation capabilities, enabling it to elicit a detectable protective immune response with only minimal quantities.53
This characteristic suggests that saRNA could be effectively applied in cancer vaccines.54 Several preclinical studies exploring this potential are already underway.
For instance, a comparative study demonstrated that saRNA targeting the human papillomavirus (HPV) exhibited stronger anti-tumor effects compared to both unmodified and modified non-replicating mRNAs.55
Despite this, it probably doesn’t work. And this paper from April cites a study that found that m1ψ replacements of uridine showed “no detectable expression”. Other noncanonical nucleotides show promise though:
However, it is currently unclear whether RNA polymerase can trigger an immune response in vivo. Furthermore, the larger size of saRNA may pose new challenges for existing delivery systems. One potential strategy to address this involves spatially segregating the sequence encoding RNA polymerase from the sequence of the target protein.
Circular RNA is another option:
Circular RNA (circRNA), with its covalent circular structure, offers enhanced stability over mRNA due to its resistance to RNase. This characteristic provides distinct advantages for RNA vaccines.56,57 The circRNA vaccine platform developed by Qu et al. has shown protective efficacy against multiple COVID-19 variants, contributing valuable insights to the development of circRNA-based cancer vaccines.58
Studies have demonstrated that circRNA molecules encoding antigens or cytokines are more effective in tumor suppression than mRNA vaccines.59,60 Additionally, Amaya et al. highlighted circRNA’s role as an adjuvant in enhancing anti-tumor effects.61 However, the precise mechanisms behind these effects are yet to be fully understood.
Cyclization: “the formation of a ring compound from a chain by the formation of a [single] new bond”.
Despite these developments, challenges remain in circRNA synthesis, including suboptimal cyclization efficiency and high costs of reagents like enzymes. Nonetheless, circRNA is emerging as a promising class of RNA-based therapeutics and vaccines with significant future potential.
They were already using dsRNA on a huge scale:
A recent advancement involves hybridizing short double-stranded RNA (dsRNA) with single-stranded mRNA to form a dendritic, comb-like structure, serving both as an antigen expression vector and immune adjuvant.
Accurate dose and quality control is virtually non-existent with the current COVID gene agents:
This method allows precise control over immune stimulation strength in mRNA vaccines by manipulating dsRNA’s length, sequence, and quantity.63 This comb-structured RNA design, applicable to various mRNA vaccines under development, is anticipated to significantly enhance their efficacy and safety.
The Next Big Thing©:
(A) Precision Medicine: Precision medicine refers to an approach that aims to tailor medical treatments and interventions to individual patients based on their specific characteristics, including their genetic makeup, environmental factors, lifestyle, and other relevant data. It emphasizes the customization of healthcare to optimize effectiveness and diminish adverse effects.
Its goal is to offer the correct treatment to the patient at the appropriate time. Precision medicine relies on advancements in technologies such as genomics, proteomics, and other "-omics" fields, as well as bioinformatics and data analysis, to identify molecular and genetic markers that can help anticipate disease risk, diagnose conditions, and guide treatment decisions (Figure 1).

From: “Precision and Personalized Medicine: Unlocking the Future of Healthcare“ (2023)
https://www.medznat.ru/en/practice/medical-billing/precision-and-personalized-medicine-unlocking-the
The flexibility of mRNA vaccine platforms is particularly valuable in personalized medicine. Customizing vaccines to match individual patient’s viral antigen profiles can address the challenge posed by the evolving nature of viral oncogenes, offering more effective treatment.
As intimated earlier, they are more hype than substance:
mRNA-based cancer vaccines that encode viral antigens
Despite the potential of mRNA vaccines to prevent viral infections linked to cancer, vaccines targeting cancer-related viruses predominantly remain in preclinical stages.
This is due to challenges such as viral mutation, immune evasion, and immune tolerance stemming from long-term infection.106–108 Future research needs to focus on overcoming these hurdles to develop more effective mRNA vaccines against these viruses.
mRNA vaccines encoding tumor antigens
Tumor-associated antigens (TAAs). TAAs are a class of antigens that are expressed at significantly higher levels in tumors compared to normal tissues. This overexpression designates them as prime targets for cancer vaccines.
As discussed, the side effects profiles of traditional therapies can be so serious that the early cessation rates can be high, and quality of life greatly reduced.
I reviewed many of much the less toxic, much less expensive, and just as efficacious alternatives here.
These vaccines, customizable based on individual TAAs, offer enhanced efficacy and reduced side effects compared to traditional therapies like chemotherapy or radiotherapy.109 This personalized approach marks a significant advancement in oncology, presenting a more targeted and patient-specific treatment strategy.
Despite their potential, mRNA vaccines targeting TAAs have limitations. TAAs, being non-mutated self-antigens, can lead to poor T cell responses and immune tolerance in clinical immunotherapy.121 Additionally, TAA expression in normal cells might cause collateral damage. These vaccines may also be less effective in cancers where TAAs have low expression levels or mutations.122
These are also largely ineffective if used on their own or before surgery, with some types of cancer. Not quite the AI powered, doctor-replacing personalised healthcare revolution that is being sold to us:
Strategies to overcome these limitations include co-delivering cancer antigens with immune-stimulatory molecules, enhancing immune activation in the tumor microenvironment, and combining vaccines with conventional treatments like chemotherapy or radiotherapy.123–126

Neoantigens are presented on the surface of cancer cells via Major Histocompatibility Complex (MHC) molecules. These neoantigens are absent in normal tissues, making them ideal targets for immunotherapeutic strategies.
As we witnessed with SARS-CoV-2 variants, you can never keep up with the mutations, and its probably the wrong approach due to the risk of recurrences if you don’t achieve 100% regression.
This is made worse if you don’t also address the underlying root causes of immune suppression and cancer pathway activation:
Their uniqueness to individual tumors circumvents the issues of tolerance and autoimmunity often associated with targeting shared tumor antigens.
In 2014, Tran et al. successfully treated a bile duct cancer patient with ERBB2IP-specific CD4+ cells, achieving complete tumor regression,127 demonstrating TSAs’ potential in inducing anti-tumor responses.
They also found gene mutations in 9 out of 10 gastrointestinal tumor patients,128 which indicates the widespread presence of tumor heterogeneity, with TSAs varying among patients even within the same tumor type.
The discussion concludes by discussing mRNA-encoded immunomodulators, such as those that can deliver tumor suppressor p53 combined with anti-PD-1 monoclonal antibodies to the immunosuppressive tumor microenvironment (TME).
We discussed at length how m1ψ can suppress the macrophage and T cell responses to the TME.
The Chinese funded review concludes by highlighting the challenges and areas for improvement. They aren’t ready yet to fulfil the vision:
Notably, the progress in personalized mRNA vaccine marks a considerable breakthrough in cancer treatment. These vaccines show enhanced potential in clinical trials, particularly when combined with existing therapies, including ICI and traditional treatments.
Despite comprehensive development, mRNA cancer vaccines still confront multiple challenges. Further research is essential to refine the development and application of these vaccines. Continued improvements in vaccine design, delivery technologies, and rapid identification of tumor neoantigens are crucial, positioning mRNA vaccines as a promising tool in personalized cancer therapy.
Conclusion
My intention was to write a Substack of further reading in support of our review. However, the wealth of research that has been done extended this work to a second post. And to be honest, the more you look, the more shocking it is. Many of these papers have been around for decades.
I wouldn’t be surprised if Karikó & Weissman were shocked too when their work came back from the dead, after effectively being dismissed for decades as too dangerous, and was seized upon as a framework for protein production and antibodies-shmantibodies at any cost. Their research went into some depth to demonstrate how just a few m1ψ nucleoside substitutions in a thousand can significantly inhibit proinflammatory immune responses.
Does Process 2 contamination repudiate our work due to IFN‐γ synthesis & CD8+ T cell stimulation? No, it doesn’t, because:
MDA5 > IFN‐γ is an inferior anti-tumor signaling pathway. Research shows that you need all the IFN pathways for a robust anti-tumor response, and for the maintenance of good health.
Our review was focused on the effects on cancer outcomes in a murine model from using m1ψ in vaccines, in the context of cancer vaccines in development and going forwards.
Further support of the hypothesis comes from the study into the effects of transfection on stromal cells signalling of part I, and also from the RIG-I KO research discussed in part II. In both cases anti-tumor immune signaling pathways were greatly suppressed, despite the presence of strong immunostimulants.
Studies show that many of the current m1ψ-modified mRNA cancer vaccines fail to reach target clinical efficacy endpoints unless you use them in combination therapy such as checkpoint inhibitors (currently a very high cost solution). Its not clear what part they actually play in outcomes, without seeing control data for comparison.
You also need to “accidentally” adulterate m1ψ-modified RNA vaccines with dsRNA, plasmid DNA or superantigenic LPS endotoxin to provoke a significant immune response. Cytotoxic cationic LNP carriers perform a similar role, with oncogenic IL-6 being significantly upregulated too.
IFN signaling pathways work together via ways we are only just beginning to understand, sometimes with almost undetectably low levels of cytokines being sufficient but essential. I’m only scratching the surface in these reviews - there is more I could discuss about IFN dysregulation, autoimmune disorders, and brain cancer. Mess with these at your peril.
The whole concept of the “100 Days Mission” to create a novel “vaccine” becomes even more laughably reckless and naïve from this angle. If someone misses a little detail, or your genetic make-up is incompatible, you may not even be aware until years later. And you will have zero chance of getting compensation once diagnosed.

DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.
References
1.
Eriksson U, Kurrer MO, Bingisser R, et al. Lethal Autoimmune Myocarditis in Interferon-γ Receptor–Deficient Mice. Circulation. 2001;103(1):18-21. doi:10.1161/01.CIR.103.1.18
2.
Xu J, Zhou Z, Zheng Y, Yang S, Huang K, Li H. Roles of inflammasomes in viral myocarditis. Front Cell Infect Microbiol. 2023;13. doi:10.3389/fcimb.2023.1149911
3.
Horwitz MS, La Cava A, Fine C, Rodriguez E, Ilic A, Sarvetnick N. Pancreatic expression of interferon-γ protects mice from lethal coxsackievirus B3 infection and subsequent myocarditis. Nat Med. 2000;6(6):693-697. doi:10.1038/76277
4.
Holm GH, Pruijssers AJ, Li L, Danthi P, Sherry B, Dermody TS. Interferon Regulatory Factor 3 Attenuates Reovirus Myocarditis and Contributes to Viral Clearance. Journal of Virology. 2010;84(14):6900-6908. doi:10.1128/jvi.01742-09
5.
Barin JG, Baldeviano GC, Talor MV, et al. Fatal Eosinophilic Myocarditis Develops in the Absence of IFN-γ and IL-17A. The Journal of Immunology. 2013;191(8):4038-4047. doi:10.4049/jimmunol.1301282
6.
Janga C, Patel T, Hennawi HA, et al. Delayed presentation of biopsy-proven eosinophilic myocarditis following COVID-19 mRNA vaccine. Glob Cardiol Sci Pract. 2023(2):e202310. doi:10.21542/gcsp.2023.10
7.
Lawson CM, Beilharz MW. Low-dose oral use of interferon inhibits virally induced myocarditis. J Interferon Cytokine Res. 1999;19(8):863-867. doi:10.1089/107999099313370
8.
Schultheiss HP, Kuhl U, Cooper LT. The management of myocarditis. European Heart Journal. 2011;32(21):2616-2625. doi:10.1093/eurheartj/ehr165
9.
On the pathogenesis of turbo cancer induced by COVID-19 mRNA vaccines: a hypothesis. Doctors for COVID Ethics. Published January 15, 2024. Accessed May 2, 2024. https://doctors4covidethics.org/on-the-pathogenesis-of-turbo-cancer-induced-by-covid-19-mrna-vaccines-a-hypothesis/
10.
Schreckenberg R, Woitasky N, Itani N, Czech L, Ferdinandy P, Schulz R. Cardiac side effects of RNA-based SARS-CoV-2 vaccines: Hidden cardiotoxic effects of mRNA-1273 and BNT162b2 on ventricular myocyte function and structure. British Journal of Pharmacology. 2024;181(3):345-361. doi:10.1111/bph.16262
11.
Tsang HW, Kwan MYW, Chua GT, et al. The central role of natural killer cells in mediating acute myocarditis after mRNA COVID-19 vaccination. Med. 2024;5(4):335-347.e3. doi:10.1016/j.medj.2024.02.008
12.
Masood KI, Yameen M, Ashraf J, et al. Upregulated type I interferon responses in asymptomatic COVID-19 infection are associated with improved clinical outcome. Sci Rep. 2021;11(1):22958. doi:10.1038/s41598-021-02489-4
13.
Knabl L, Lee HK, Wieser M, et al. BNT162b2 vaccination enhances interferon-JAK-STAT-regulated antiviral programs in COVID-19 patients infected with the SARS-CoV-2 Beta variant. Commun Med. 2022;2(1):1-10. doi:10.1038/s43856-022-00083-x
14.
Deng Y, Fu H, Han X, et al. Activation of DDX58/RIG‑I suppresses the growth of tumor cells by inhibiting STAT3/CSE signaling in colon cancer. Int J Oncol. 2022;61(4):120. doi:10.3892/ijo.2022.5410
15.
Cao S, Long X, Xiao L, et al. DDX58 deficiency leads to triple negative breast cancer chemotherapy resistance by inhibiting Type I IFN-mediated signalling apoptosis. Front Oncol. 2024;14. doi:10.3389/fonc.2024.1356778
16.
Jiang S, Deng X, Luo M, et al. Pan-cancer analysis identified OAS1 as a potential prognostic biomarker for multiple tumor types. Front Oncol. 2023;13:1207081. doi:10.3389/fonc.2023.1207081
17.
Jia H, Liu X, Wang Z, Zhang W, Chen X. A Pan-Cancer Analysis Reveals OAS2 as a Biomarker for Cancer Prognosis and Immunotherap. Published online 2023. doi:10.21203/rs.3.rs-3097514/v1
18.
Li X yu, Hou L, Zhang L yu, et al. OAS3 is a Co-Immune Biomarker Associated With Tumour Microenvironment, Disease Staging, Prognosis, and Treatment Response in Multiple Cancer Types. Front Cell Dev Biol. 2022;10:815480. doi:10.3389/fcell.2022.815480
19.
Fairweather D, Frisancho-Kiss S, Yusung SA, et al. Interferon-γ Protects against Chronic Viral Myocarditis by Reducing Mast Cell Degranulation, Fibrosis, and the Profibrotic Cytokines Transforming Growth Factor-β1, Interleukin-1β, and Interleukin-4 in the Heart. Am J Pathol. 2004;165(6):1883-1894. Accessed May 3, 2024. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1618717/
20.
Buskiewicz IA, Huber SA. Mda5. Circulation: Heart Failure. 2013;6(2):153-155. doi:10.1161/CIRCHEARTFAILURE.113.000137
21.
Ristic AD, Maisch B. Cardiac rhythm and conduction disturbances: what is the role of autoimmune mechanisms? Herz. 2000;25(3):181-188. doi:10.1007/s000590050005
22.
STAT1. In: Wikipedia. ; 2024. Accessed May 24, 2024. https://en.wikipedia.org/w/index.php?title=STAT1&oldid=1212657316
23.
Chromatin. Accessed May 24, 2024. https://www.genome.gov/genetics-glossary/Chromatin
24.
Kim H, Ahn HS, Hwang N, et al. Epigenomic landscape exhibits interferon signaling suppression in the patient of myocarditis after BNT162b2 vaccination. Sci Rep. 2023;13:8926. doi:10.1038/s41598-023-36070-y
25.
Coux RX, Owens NDL, Navarro P. Chromatin accessibility and transcription factor binding through the perspective of mitosis. Transcription. 2020;11(5):236. doi:10.1080/21541264.2020.1825907
26.
Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718-724. doi:10.1126/science.abc6027
27.
Eskandarian Boroujeni M, Sekrecka A, Antonczyk A, et al. Dysregulated Interferon Response and Immune Hyperactivation in Severe COVID-19: Targeting STATs as a Novel Therapeutic Strategy. Front Immunol. 2022;13:888897. doi:10.3389/fimmu.2022.888897
28.
IRF3. In: Wikipedia. ; 2023. Accessed May 24, 2024. https://en.wikipedia.org/w/index.php?title=IRF3&oldid=1189310515
29.
Rinchai D, Deola S, Zoppoli G, et al. High–temporal resolution profiling reveals distinct immune trajectories following the first and second doses of COVID-19 mRNA vaccines. Science Advances. 2022;8(45):eabp9961. doi:10.1126/sciadv.abp9961
30.
Roh YS, Park S, Kim JW, Lim CW, Seki E, Kim B. Toll-like receptor 7-mediated type I interferon signaling prevents cholestasis- and hepatotoxin-induced liver fibrosis. Hepatology. 2014;60(1):237-249. doi:10.1002/hep.26981
31.
Zhang X, Wang S, Zhu Y, et al. Double-edged effects of interferons on the regulation of cancer-immunity cycle. Oncoimmunology. 10(1):1929005. doi:10.1080/2162402X.2021.1929005
32.
Wittling MC, Cahalan SR, Levenson EA, Rabin RL. Shared and Unique Features of Human Interferon-Beta and Interferon-Alpha Subtypes. Front Immunol. 2021;11:605673. doi:10.3389/fimmu.2020.605673
33.
Pezzullo AM, Axfors C, Contopoulos-Ioannidis DG, Apostolatos A, Ioannidis JPA. Age-stratified infection fatality rate of COVID-19 in the non-elderly population. Environ Res. 2023;216:114655. doi:10.1016/j.envres.2022.114655
34.
Li C, Lee A, Grigoryan L, et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat Immunol. 2022;23(4):543-555. doi:10.1038/s41590-022-01163-9
35.
Jin R, Cao X, Lu M, Gao Q, Ma T. The intersection molecule MDA5 in Cancer and COVID-19. Front Immunol. 2022;13:963051. doi:10.3389/fimmu.2022.963051
36.
Nair A, Jacob S. A simple practice guide for dose conversion between animals and human. Journal of Basic and Clinical Pharmacy. 2016;7:27-31. doi:10.4103/0976-0105.177703
37.
Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology. 2017;6(5):e1314424. doi:10.1080/2162402X.2017.1314424
38.
Verbeke R, Hogan MJ, Loré K, Pardi N. Innate immune mechanisms of mRNA vaccines. Immunity. 2022;55(11):1993. doi:10.1016/j.immuni.2022.10.014
39.
Milano G, Gal J, Creisson A, Chamorey E. Myocarditis and COVID-19 mRNA vaccines: a mechanistic hypothesis involving dsRNA. Future Virol.:10.2217/fvl-2021-0280. doi:10.2217/fvl-2021-0280
40.
Apostolidi M, Stamatopoulou V. Aberrant splicing in human cancer: An RNA structural code point of view. Front Pharmacol. 2023;14:1137154. doi:10.3389/fphar.2023.1137154
41.
Dias Junior AG, Sampaio NG, Rehwinkel J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019;27(1):75-85. doi:10.1016/j.tim.2018.08.007
42.
Iurescia S, Fioretti D, Rinaldi M. The Innate Immune Signalling Pathways: Turning RIG-I Sensor Activation against Cancer. Cancers (Basel). 2020;12(11):3158. doi:10.3390/cancers12113158
43.
Poeck H, Wintges A, Dahl S, Bassermann F, Haas T, Heidegger S. Tumor cell-intrinsic RIG-I signaling governs synergistic effects of immunogenic cancer therapies and checkpoint inhibitors in mice. European Journal of Immunology. 2021;51(6):1531-1534. doi:10.1002/eji.202049158
44.
Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167(3):1179-1187. doi:10.4049/jimmunol.167.3.1179
45.
Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721-728. doi:10.1007/s00262-004-0653-2
46.
Janga C, Patel T, Hennawi HA, et al. Delayed presentation of biopsy-proven eosinophilic myocarditis following COVID-19 mRNA vaccine. Glob Cardiol Sci Pract. 2023(2):e202310. doi:10.21542/gcsp.2023.10
47.
Guo R, Lu SY, Ma JX, et al. RIG-I acts as a tumor suppressor in melanoma via regulating the activation of the MKK/p38MAPK signaling pathway. Hum Cell. 2022;35(4):1071-1083. doi:10.1007/s13577-022-00698-1
48.
Yao R, Xie C, Xia X. Recent progress in mRNA cancer vaccines. Hum Vaccin Immunother. 20(1):2307187. doi:10.1080/21645515.2024.2307187
49.
After coronavirus vaccine breakthrough, mRNA pioneers pivot to cancer. POLITICO. Published January 24, 2022. Accessed May 29, 2024. https://www.politico.eu/article/mrna-cancer-vaccine-coronavirus/
50.
Semmler A, Mundorf AK, Kuechler AS, et al. Chronic Fatigue and Dysautonomia following COVID-19 Vaccination Is Distinguished from Normal Vaccination Response by Altered Blood Markers. Vaccines (Basel). 2023;11(11):1642. doi:10.3390/vaccines11111642
51.
Performance Status or Fatigue (St George’s Acute Oncology Service). Accessed May 29, 2024. https://www.stgeorges.nhs.uk/aos/community-healthcare-professional/guidance/performance-status-or-fatigue/
52.
Parhiz H, Brenner JS, Patel PN, et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J Control Release. 2022;344:50-61. doi:10.1016/j.jconrel.2021.12.027
53.
Definition of neoantigen - NCI Dictionary of Cancer Terms - NCI. Published February 2, 2011. Accessed May 30, 2024. https://www.cancer.gov/publications/dictionaries/cancer-terms/def/neoantigen
54.
Azizi H, Renner TM, Agbayani G, et al. Self-amplifying RNAs generated with the modified nucleotides 5-methylcytidine and 5-methyluridine mediate strong expression and immunogenicity in vivo. NAR Molecular Medicine. 2024;1(2):ugae004. doi:10.1093/narmme/ugae004
55.
Zhong Y, Du Y, Yang X, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Molecular Cancer. 2018;17(1):79. doi:10.1186/s12943-018-0827-8
56.
Yao H, Dittmann M, Peisley A, et al. ATP-dependent effector-like functions of RIG-I like receptors. Mol Cell. 2015;58(3):541-548. doi:10.1016/j.molcel.2015.03.014
Hat tip to Genervter: "Pancreatic vaccines with amazing results".
Investigational mRNA Vaccine Induced Persistent Immune Response in Phase 1 Trial of Patients With Pancreatic Cancer
April 7, 2024
https://www.mskcc.org/news/can-mrna-vaccines-fight-pancreatic-cancer-msk-clinical-researchers-are-trying-find-out
***
Observations. And this is a recurrent theme:
1/ It's a phase 1 trial, which is primarily to assess safety and side effects, not efficacy.
2/ It's no cure and again after surgery, supposedly to improve survival rates. With pancreatic cancer this excludes a lot of patients as only a small number are suitable for surgery. It's aggressive, and with most it's already too late and metastatic once diagnosed.
3/ It's ineffective on its own and was used in combination with an immunotherapy drug called atezolizumab and a chemotherapy regimen called mFOLFIRINOX. The irony here is that a common side effect of atezolizumab is... drumroll... pancreatitis!
https://www.mskcc.org/cancer-care/patient-education/medications/adult/atezolizumab
4/ Even so, the efficacy was at or below 50%.
If only say 20% are suitable for surgery and the efficacy in this group is 50% then the overall contribution to improving pancreatic cancer survival rates is probably less than 10%.
And I'm being generous here, as only around half the patients are suitable for chemo:
"... The main concern regarding adjuvant chemotherapy is that half of the patients never start adjuvant treatment as a result of surgical complications, clinical deterioration, or early recurrence [6, 7]. "
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10405377/
Is it worth the horrible side effects, expense, and loss of QoL to improve your odds a little?
5/ What sort of T cell response? It may have little effect.
6/ Pancreatic cancer involves so many genetic mutations and disrupted cancer pathways that a needle just isn't going to put Humpty back together again:
Genetic Mutations of Pancreatic Cancer and Genetically Engineered Mouse Models
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8750056/
If TLR2 or 4 recognise the dsRNA you used, then you may accelerate tumor progression:
Immunomodulatory Effects of dsRNA and Its Potential as Vaccine Adjuvant (2010)
https://www.hindawi.com/journals/bmri/2010/690438/