


Self-amplifying mRNA (saRNA) Part 3, Episode VI: Return of the Cancer
"In order to build your immunity, we need to destroy it"
This Substack is too long for email. Please click on the title to load in the app or a browser.
Reading time:
short story - novelette - novella - novel - PhD thesis - War and Peace - U.S. Tax Code
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.


Contents

Introduction
I originally expected to write just the two Substacks to say the quiet part out loud about saRNA tech. Needless to say, once you start going down the rabbit hole you uncover a lot more pathologies that warrant consideration.
To keep within a self-imposed 10,000 word limit necessitates serialising the discussion, much as I needed to do when writing about the “essential giant mineral” magnesium in 2024.
The story so far:
“Self-amplifying mRNA (saRNA) Part 1: The Japanese registration documents” was a translation and annotation of Dr Stebel’s excellent walkthrough of the Japanese (pre-) registration documents for ARCT-154 (“Kostaive”).
“Self-amplifying mRNA (saRNA) Part 2, Episode V: The Pharma Empire Strikes Back” discussed how vaccinia virus immune evasion proteins E3, K3, and B18 act together synergistically as “a highly potent blocker of PKR activation and of interferon (IFN)-β upregulation.”
This Substack will continue the discussion, investigating the role of each innate inhibiting protein (IIP) in disease, with a focus on cancer mechanisms.
Discussion
Cancer pathways
These particular IIPs are subject to patent (part 4) and may not be in every commercial product, but suppression of innate immune mechanisms is essential, using whatever means are necessary. This is to try to reduce side effects and enhance protein synthesis and duration when playing the antibody game.
- PKR
PKR: protein kinase R, is an interferon-induced, double-stranded RNA-activated protein kinase that is activated by viral infections and inhibits viral protein synthesis.
It has two roles in cancer, but it is context-sensitive, depending on cancer type. It may act as a tumour suppressor for colorectal cancer (CRC) and other p53-associated cancers. More than 50% of human primary tumours are associated with p53 dysfunction, usually due to inactivation of the TP53 gene due to missense mutations.
Examples of p53-associated cancers include sarcomas, brain tumours, adrenal cortical carcinomas, ovarian cancer, breast cancer, colorectal cancer, head and neck cancer, bladder, haematopoietic and lung cancer1.
Key takes from “PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53” by Yoon et al. (2009)2:
Here, we report that PKR is a p53 target gene and plays an important role in the tumor-suppressor function of p53.
Activation of p53 by genotoxic stress induces a significant level of PKR expression by acting on the newly identified cis-acting element (ISRE), which is separated from the IFN-stimulated responsive element on the PKR promoter, resulting in translational inhibition and cell apoptosis.
The genotoxin-mediated inhibition of translation is associated with the p53/PKR/eIF2a (eukaryotic initiation factor-2α) pathway.
To some extent, p53 activation induced by DNA damage facilitates cell apoptosis by activating PKR.
PKR-knockdown human colon cancer cells grew rapidly in nude mice and proved resistant to anti-cancer drugs. These data indicate that p53-mediated tumor suppression can be attributed at least in part to the biological functions of PKR induced by p53 in genotoxic conditions.
However, for other cancers it leads to a worse prognosis and progression. High PKR expression in CD34+ cells of acute myeloid leukaemia (AML) patients correlates with worse survival and shortened remission duration, because it inhibits DNA damage response signalling and double-strand break repair, leading to the accumulation of potentially oncogenic mutations3.
The negatives appear to outweigh the positives. From 2018, “Roles of protein kinase R in cancer: Potential as a therapeutic target” by Watanabe et al4. discusses its role in malignant diseases:
It was initially identified as an innate immune antiviral protein induced by interferon (IFN) and activated by dsRNA. PKR is recognized as a key executor of antiviral host defense. Moreover, it contributes to inflammation and immune regulation through several signaling pathways.
In addition to IFN and dsRNA, PKR is activated by multiple stimuli and regulates various signaling pathways including the mitogen‐activated protein kinase (MAPK) and nuclear factor kappa‐light‐chain‐enhancer of activated B cells pathways.
PKR was initially thought to be a tumor suppressor as a result of its ability to suppress cell growth and interact with major tumor suppressor genes.
However, in several types of malignant disease, such as colon and breast cancers, its role remains controversial. In hepatocellular carcinoma, hepatitis C virus (HCV) is the main cause of liver cancer, and PKR inhibits HCV replication, indicating its role as a tumor suppressor.
However, PKR is overexpressed in cirrhotic patients, and acts as a tumor promoter through enhancement of cancer cell growth by mediating MAPK or signal transducer and activator of transcription pathways.
Moreover, PKR is reportedly required for the activation of inflammasomes and influences metabolic disorders.


As with many diseases, an imbalance in PKR levels caused by vaccination may disrupt homeostasis, leading to unpredictable outcomes.


- K3
Via another part of the PKR pathway, K3: a vaccinia virus-encoded eIf-2 alpha homolog (mimic), abrogates the antiviral effects of interferon by reducing the level of eIf-2 alpha phosphorylation5.
A paper from 1997 by Sharp et al. discusses the mechanisms.
Explain like I’m 5 (ELI5): The K3 IIP mimics part of antiviral eIf-2 alpha, binds with part of its target protein, PKR, and stops it from being activated by phosphorylation. If PKR was acting as a tumour suppressor, then hold my beer:
Key takes from “Homologous regions of the alpha subunit of eukaryotic translational initiation factor 2 (eIF2alpha) and the vaccinia virus K3L gene product interact with the same domain within the dsRNA-activated protein kinase (PKR)”6
The vaccinia virus K3L gene product, pK3, binds to the dsRNA-activated protein kinase, PKR, reducing its ability to interact with and phosphorylate eIF2alpha.
On the basis of this characteristic and the homology of pK3 to the N-terminus of eIF2alpha, several laboratories have utilized pK3 to investigate the molecular determinants that specify substrate recognition by PKR.
The data presented here demonstrate that the natural substrate, eIF2alpha, also binds to PKR in vitro and interacts with the same or an overlapping domain within PKR.
A truncated form of eIF2alpha, representing the N-terminal 123 amino acids and containing the regions of homology to pK3, retains the ability to bind PKR. pK3, eIF2alpha, and the truncated form of eIF2alpha all bind to the C-terminus of PKR containing the catalytic domain, but not to the regulatory N-terminus.
Variants of pK3 and eIF2alpha, des-(75-78)-K3L (pK3deltaGYID), and des-(80-83)-eIF2alpha (eIF2alphadeltaGYID), from which the conserved amino acids GYID have been deleted, exhibit a decreased ability to interact with PKR.
Similarly, the in vitro binding of pK3, eIF2alpha, and the truncated form of eIF2alpha to PKR can be competed with purified pK3 but not with pK3deltaGYID. In addition, the deletion of GYID from eIF2alpha significantly reduces its ability to be phosphorylated by PKR, demonstrating that PKR recognizes its substrate, at least in part through interaction with sequences remote from the phosphorylation site.
In summary, we have shown that the region within PKR that interacts with the pseudosubstrate, pK3, is the same region that interacts with the authentic substrate, eIF2alpha. In addition, we have shown that the N-terminal 123 amino acids of eIF2alpha contains structural elements necessary for recognition by PKR.
The results pinpoint the GYID motif, shared between pK3 and eIF2alpha and distant from the phosphorylation site, as being important for the interaction of eIF2alpha with PKR, as well as its phosphorylation.
The K3L gene product inhibits PKR activity both in vitro and in vivo [39-421. Furthermore, pK3 is able to down regulate two other eIF2a kinases, HRI, the heme-sensitive kinase, and GCN2, the amino-acid-starvation responsive kinase of yeast [9, 40, 42, 43].
This suggests that all three kinases share structural features that allow them to recognize eIF2a as a substrate. The K3L gene product interacts directly with PKR in vitro as revealed by co-immunoprecipitation using anti-PKR antibody [40,41].
In addition, pK3 also interacts with PKR in vivo, as deter-mined by co-localization with PKR in vaccinia-virus-infectedcells [41], as well as by use of the yeast two-hybrid system [34-36].
Given the sequence similarity, together with its ability to bind to PKR and prevent eIF2a phosphorylation, pK3 is considered to function as a pseudosubstrate [39-41, 43].
In addition, our results show that a region of similarity, shared between pK3 and eIF2a, is required for both proteins to bind stably to PKR, and its deletion significantly compromises the ability of PKR to phosphorylate eIF2a.
Earlier studies suggested that pK3 has a higher affinity for PKR than eIF2a, based on the observation that purified eIF2 heterotrimer does not compete with "S-labeled pK3 for PKR binding [41]. Based on these observations, the pseudosubstrate pK3 has been increasingly used as a tool for studying PKR and related eIF2a protein kinases[34-36, 40-43].
Why is the reduction of phosphorylated eIF2α levels by K3 a problem? An in vitro study by Guo et al. from 2017 used a tissue microarray containing 233 tumours from breast cancer patients to screen for p-eIF2α. They found that the more p-eIF2α the patient has, the better the prognosis.
Their findings are described in the title: “Phosphorylated eIF2α predicts disease-free survival in triple-negative breast cancer patients”7
Key takes:
Phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α), which functions as a marker of endoplasmic reticulum stress, has been reported to be associated with patient prognosis in various cancers.
However, little is known about the prognostic value of p-eIF2α in breast cancer, particularly in different breast cancer subtypes.
An immunohistochemistry screen for p-eIF2α was performed using a tissue microarray containing 233 tumors and paired peritumoral tissues from female patients diagnosed with breast cancer.
The staining results were scored semiquantitatively, and the p-eIF2α expression level in breast cancer and its potential prognostic value were investigated.
In this retrospective cohort study, we found that p-eIF2α levels were significantly upregulated in breast cancer (P < 0.001).
p-eIF2α level was negatively correlated with lymph node status (P = 0.039).
Survival analysis by Kaplan–Meier estimation and Cox regression showed that p-eIF2α level was correlated with better disease free survival (P = 0.026) and served as an independent prognostic factor (P = 0.046) in patients with triple-negative breast cancer.
Our study revealed that p-eIF2α was upregulated in breast cancer and represented a novel predictor of prognosis in patients with triple-negative subtype.
Important note: Phosphorylation of eIF2 in cancer is via the pancreatic eIF2α kinase PEK or PERK pathway, and vaccinia virus K3L is a demonstrated inhibitor of each of the known eIF2α kinases8:
The endoplasmic reticulum (ER) is a eukaryotic cell organelle responsible for lipid biosynthesis, intracellular Ca2+ homeostasis, and protein folding and transport. The ER protein-folding environment can be disrupted by numerous events, including nutrient fluctuations, as well as environmental, physiological, and pathological damage.
The protein misfolding and accumulation that results from such disruption is termed ER stress1. The unfolded protein response (UPR) is a collection of signaling pathways that respond to unfolded proteins in the ER lumen2.
This links back to the recent Substack about vaccine-induced HSP autoAbs:
Extensive evidence suggests that ER stress and UPR activation are involved in the development of several cancer types and play important roles in every aspect of cancer, including tumor initiation, development and progression.
The UPR comprises three principal parallel branches: the PKR-like ER kinase (PERK)–eukaryotic translation initiation factor (eIF) 2α pathway; the inositol-requiring protein 1α (IRE1α)–X-box binding protein 1(XBP1) pathway; and the activating transcription factor (ATF) 6α pathway3.
Elevated levels of phosphorylated eIF2α may also be associated with cancer progression:
The function of the PERK–eIF2α pathway in tumors is still uncertain; signaling via this pathway may induce either survival or apoptosis of tumor cells upon ER stress, and can either promote or inhibit malignant transformation.
eIF2 comprises three subunits: α, β, and γ. The α-subunit of eIF2, eIF2α, can be phosphorylated on Ser51, thereby effectively reducing the level of active eIF2.
In this way, p-eIF2α can significantly inhibit mRNA translation initiation8 and global protein synthesis9.
Recently, evidence has mounted to demonstrate that p-eIF2α upregulation is associated with tumor development and progression10,11,12.
Conversely, other studies have demonstrated that p-eIF2α has a potential protective effect13,14,15. Thus, the detailed functions of p-eIF2α in tumors remain unclear.

Elevated p-eIF2α was associated with a much greater risk of recurrence of pancreatic cancer:
High level of p-eIF2α expression correlated with significantly higher risk of recurrence/metastasis after surgery. Among the 47 patients whose tumors were p-eIF2α-high, 38 (80.9%) had recurrence/metastasis compared to 54.1% (20/37) in those whose tumors were p-eIF2α-low (P = 0.02).
From: “Expression and Clinical Significance of PERK and Phosphorylated EIF2α in Pancreatic Ductal Adenocarcinoma“ (2020)
Conversely, high levels of phosphorylated p-eIF2α are strongly associated with disease-free survival (DFS) with some cancer types, such as breast cancer:

The effects are particularly striking with triple-negative breast cancer (TNBC).
If you are a TNBC patient then I would strongly advise avoiding being boosted with saRNA products like the, er, plague, as there may be a 40% chance of you being dead in as little as 4 years:

As for other cancers, we just don’t know yet:
The prognostic value of p-eIF2α in tumor types other than breast cancer is still unclear, as described above. p-eIF2α inhibits the synthesis of large amounts of proteins that is a necessary part of the tumorigenesis process.
However, it may lead to an increase in ATF4, CHOP and other factors23,24, all of which may aid or impede tumor progression depending on the extent of the stress.
We suggest that p-eIF2α has a predominantly inhibitory effect on tumor growth in TNBC. As we expected, higher p-eIF2α expression was associated with lower tumor invasion of lymph nodes.
Of the various breast cancer subtypes, TNBC has the greatest need for improved therapies because it is clinically aggressive and usually relapses and progresses in a short time.
Although sensitive to conventional chemotherapy25, TNBC remains the breast cancer subtype with the worst patient prognosis.
Moreover, TNBC therapy remains challenging because of the underlying heterogeneity of TNBC and the lack of predictive biomarkers and effective therapeutic targets.
In this study, we illustrated that p-eIF2α may be a potential target for the treatment of TNBC.
Aktas et al. previously identified three small molecular weight compounds that induce eIF2α phosphorylation, for use in cancer therapy26. However, therapeutic targeting of p-eIF2α remains challenging because of the dual function of p-eIF2α, which could result in severe side effects.
In this study, we demonstrated that p-eIF2α predicted disease-free survival and could serve as an independent prognostic biomarker in TNBC. This finding suggests that evaluating p-eIF2α expression in breast cancer may have a potential clinical application, by providing additional information for oncologists when individualizing cancer management.
Keeping your engineered RNA product shareholders happy by suppressing IFN signalling pathways is not without consequences, and long-term outcomes should not be down to luck alone. These products need to be restricted pending further investigations.

- MERS-CoV ORF4a
ORF: Open reading frame, a DNA sequence that starts with a start codon and ends with a stop codon, and has the potential to be translated into a polypeptide.
Of interest here is that this IIP is derived from MERS - Middle East Respiratory Syndrome. MERS is almost certainly the result of gain of function work. After being confined to bat colonies for countless millennia it somehow managed to make two zoonotic jumps - from bats to camels, and camels to humans, first officially appearing in a human in 2012.
The GOF work would have been on its fusion cleavage site, so that it could become more virulent and infect human endothelial cells.
Yes, we know the “proximal origin” draft was a work of fiction to distract and divert, but their discussion about the FCS is interesting:
February 4, 2020
Edward Holmes emails the first draft of proximal origin to Jeremy Farrar with the comment: "It's fundamental science and completely neutral as written. Did not mention other anomalies as this will make us look like loons."
Farrar emails the redraft to Fauci and Collins with the note: "Please treat in confidence - a very rough first draft from Eddie and team - they will send on the edited, cleaner version later. Pushing WHO again today."
The early draft states that furin cleavage sites can arise in betacoronaviruses in the lab through serial passage. The citation: A call in collaboration with the National Academies of Sciences, Engineering, and Medicine. It’s not precisely clear who stated that betacoronaviruses could acquire a furin cleavage site in serial passage, but Andersen was one of just eight experts tapped by NASEM. Two of the other experts were EcoHealth Alliance President Peter Daszak and University of North Carolina virologist Ralph Baric. Thus the early draft described serial passage in the lab as one of the ways the furin cleavage site might have arisen. “Basic research involving passage of bat SARS-like coronaviruses in tissue culture and/or animal models have been ongoing in BSL-2 for many years across the world, including in Wuhan,” the draft reads.
On the importance of its FCS:
The emergence of Middle East respiratory syndrome coronavirus (MERS-CoV), a deadly human coronavirus, has triggered considerable interest in the biomedical community. Similar to other enveloped viruses, coronaviruses access host cells by membrane fusion, a process mediated by specific fusion or “spike” proteins on the virion, often activated by cellular proteases. We have identified unique features of the MERS-CoV spike (S) protein cleavage activation. Our findings suggest that S can be activated by furin, a broadly expressed protease, by a two-step cleavage mechanism, occurring at distinct sites, with cleavage events temporally separated. Such furin-mediated activation is unusual in that it occurs in part during virus entry.
From: “Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein” (2014)
In effect, the proposal is to use a protein from one lab-derived virus to try to help build immunity to a disease caused by another lab-derived virus.
The problem with using ORF4a as an IIP is that it significantly downregulates the levels of a pattern recognition receptor (PRR) protein called melanoma differentiation-associated gene 5 (MDA5)9, which has a role in both innate immunity and cancer surveillance.
Key takes from “The intersection molecule MDA5 in Cancer and COVID-19” (2022) by Jin et al.10:
Cytoplasmic nucleic acid sensor melanoma differentiation-associated gene 5 (MDA5) serves as an important pattern recognition receptor in the innate immune system by recognizing viral RNA.
MDA5 also plays a role in identifying the cytoplasmic RNA from damaged, dead cancer cells or autoimmune diseases.
MDA5’s recognition of RNA triggers innate immune responses, induces interferon (IFN) response and a series of subsequent signaling pathways to produce immunomodulatory factors and inflammatory cytokines.
The RNA vaccine mimics the virus:
Latest data show that in COVID-19, SARS-CoV-2 replicates in lung epithelial cells and induces delayed interferon (IFN) response, in which MDA5 acts as the main sensor to recognize SARS-CoV-2 infection and trigger antiviral response (18).
Unfortunately, viruses can either disturb key regulatory structures, or interfere with interferon (IFN) system in multiple ways, so as to escape the body’s defense mechanism.
MDA5 gene was first reported to be induced by differentiation, cancer reversal and apoptosis in 2002 (30).
MDA5 and cancer:
Tumor cells share common key characteristics, such as oxidative stress, genomic instability and mutation, and changes in metabolic rate, which can lead to nuclear or mitochondrial DNA damage, thus releasing damaged nucleic acid fragments into the cytoplasm (42).
In mammalian cells, two typical cytoplasmic nucleic acid sensing pathways are cGAS-stimulator of interferon genes (STING) and RLRs-MAVS pathways, which are responsible for the recognition of cytoplasmic DNA and RNA, respectively (43, 44).
MDA5 as cytoplasmic dsRNA sensor recognizes long-chain high-order dsRNA structures (45). The MDA5 signaling pathway mainly interacts with the adapter MAVS and recruits TBK1. The phosphorylation cascade allows signal transduction to lead to the production of IRF3 and IRF7, which usually leads to the production of IFNs, which subsequently induces the activation of ISGs and NF-κB target genes ( Figure 1 ) (20).
Left panel: Non-structural proteins from viruses, such as ORF4a, inhibit MDA5. Engineered saRNA and m1u-modified mRNA agents have the same effect. nsP4 comes from the ORF4 gene11.
Right panel: MDA5 inhibits cancer by causing programmed cell death (apoptosis) or by the IFN type 1 anti-tumour pathways:

A frequently used cancer treatment mimics a viral infection to trigger the MDA5 pathway:
The most widely used tumor treatment strategy is the use of anti-tumor DNA demethylating agents and DNA methyltransferase inhibitors (DNMTis) (46, 47). Demethylation in the normal suppressed region of the genome can lead to endogenous retrovirus transcription, triggering cytoplasmic dsRNA sensing in cancer cells to activate MDA5 and MAVS, resulting in reduced cell growth and self-renewal, thus mimicking viral infection ( Figure 1 ) (48, 49).
NSs: nucleic acid sensors. These may turn a cold tumour hot, but you need to target the right PRRs or you may have the opposite effect. This is something I discussed in the IgG4 series of Substacks.
Surprisingly, the activation of NSs can change the tumor microenvironment (TME) from immunosuppressive state to pro-inflammatory state (52–54).
However, some studies have shown that PRRs and IFN signals promote tumor progression or immune tolerance, but not pro-inflammatory effect in immune cells (55), indicating that we need to target the PRRs of immune cells in TME in the future to improve the efficacy.
Therefore, the CAR-T cells were engineered to produce a non-coding RNA called ‘RN7SL1’ as a new PRR agonist ( Figure 1 ). Normally, RN7SL1 can be shielded from being recognized by PRR, but when it is not shielded, it can mimic viral RNA and activate nucleic acid sensors MDA5 and RIG-I, thus enhancing endogenous anti-tumor immunity (54, 56).
Great in theory, but chimeric antigen receptor (CAR)-T cell therapies are an example of what can go wrong:
Chimeric antigen receptor (CAR)-T cell therapies have always carried a risk of causing secondary malignancies, but until now that risk has been somewhat theoretical. In the last two months, since the FDA launched an investigation into the risk of developing secondary cancers following the administration of CAR-T cell therapies, a volley of safety updates has increased the scrutiny of these innovative therapies.
One month after the FDA opened its investigation, Johnson and Johnson and Legend Biotech reported data from the Phase II CARTITUDE-1 trial (NCT03548207) showing that myeloid neoplasms occurred in 10 out of 97 patients following treatment with its CAR-T cell therapy Carvykti (ciltacabtagene autoleucel; cilta-cel). Nine of these 10 patients died after developing myeloid neoplasms. In December, the companies added a boxed warning stating the risk of developing secondary hematological malignancies following treatment with Carvykti.
Then, on 19 January, the FDA sought to add boxed warnings for secondary T-cell malignancies to all approved CAR-T therapies, including Carvykti. A few days later, the agency updated the proposed label for Gilead Sciences’ CAR-T treatment Tecartus (brexucabtagene autoleucel) to a general class-wide boxed warning instead of the specific warning that was previously issued.
From “CAR-T therapies and cancer risk: No easy answers for the FDA” (January 2024)
Since MDA5 dominates the perception of synthetic dsRNA analogue poly (I:C) and triggers the cytoplasmic interferon response ( Figure 1 ).
Another link between innate immunity and TNBC:
Studies on triple negative breast cancer (TNBC) have shown that Poly I:C inhibits transforming growth factor-β (TGF-β) signal transduction in a MDA5 or RIG-I-dependent manner, thus promoting cancer cell death, and this effect can be weakened by forced expression of Smad3 (57).
Since TGF- β is the characteristic of promoting the migration, invasion, bone metastasis and survival of tumor cells in TNBC, inhibition of TGF-β may be an effective strategy for the treatment of metastatic cancer.
Interestingly, spliceosome-targeted therapies can also trigger an antiviral immune response in TNBC through dsRNA formation from mis-spliced RNA ( Figure 1 ) (58).
MDA5 and suppression of malignant melanoma:
In melanoma cells, RIG-I and MDA5 also initiate a p53-independent Noxa pro-apoptotic signal pathway, which is independent of type I IFN response (61, 66, 67).
Although this pro-apoptotic signal pathway is also active in non-malignant cells, the sensitivity of these cells to apoptosis is much lower than that of melanoma cells.
Bcl-xL is an anti-apoptotic protein. Expression is increased in primary colorectal adenocarcinomas12, and it also blocks MDA5-mediated apoptosis:
Endogenous Bcl-xL can block RIG-I and MDA5-mediated apoptosis in non-malignant cells.
Both RIG-I and MDA5 ligands can also reduce the lung metastasis of human tumor in immunodeficient mice. These results confirm that RIG-I and MDA5 ligands have therapeutic potential in solid tumors through inducing tumor cells to apoptosis.
In general, targeting MDA5 signaling pathway has promising advantages in cancer immunotherapy.

Innate immunity and cancer
Tumour suppression isn’t just due to one innate immunity element but is the product of several mechanisms and pathways working together.
This is a more holistic approach to cancer prevention, something that relates more to diet and lifestyle. It’s an area that allopathic medicine often fails to address due to its focus on single pathways and of using single, patented molecules to attempt to treat cancer.
The immune system can identify and destroy nascent tumor cells in a process termed cancer immunosurveillance, which functions as an important defense against cancer. Recently, data obtained from numerous investigations in mouse models of cancer and in humans with cancer offer compelling evidence that particular innate and adaptive immune cell types, effector molecules, and pathways can sometimes collectively function as extrinsic tumor-suppressor mechanisms.
From: “Natural Innate and Adaptive Immunity to Cancer“ (2011)
https://www.annualreviews.org/content/journals/10.1146/annurev-immunol-031210-101324
Innate immune responses are typically associated with improved prognoses for cancer patients, and the inverse is true too.
Key takes from “Innate Immune Recognition of Cancer” by Woo et al. (2015)13:
The observation that a subset of cancer patients show evidence for spontaneous CD8+ T cell priming against tumor-associated antigens has generated renewed interest in the innate immune pathways that might serve as a bridge to an adaptive immune response to tumors.
Manipulation of this endogenous T cell response with therapeutic intent—for example, using blocking antibodies inhibiting PD-1/PD-L1 (programmed death-1/programmed death ligand 1) interactions—is showing impressive clinical results. As such, understanding the innate immune mechanisms that enable this T cell response has important clinical relevance.
Defined innate immune interactions in the cancer context include recognition by innate cell populations (NK cells, NKT cells, and γδ T cells) and also by dendritic cells and macrophages in response to damage-associated molecular patterns (DAMPs).
Recent evidence has indicated that the major DAMP driving host antitumor immune responses is tumor-derived DNA, sensed by the stimulator of interferon gene (STING) pathway and driving type I IFN production. A deeper knowledge of the clinically relevant innate immune pathways involved in the recognition of tumors is leading toward new therapeutic strategies for cancer treatment.
This review summarizes the current state of knowledge regarding innate cell populations, as well as sensing mechanisms relevant for the cancer context, which is typically sterile, without obvious pathogen involvement.
Emphasis is placed on recent data implicating host type I IFN production as a key innate immune component and the role of the stimulator of interferon gene (STING) pathway of cytosolic DNA sensing in driving type I IFN production and adaptive immunity.
INNATE IMMUNE CELL POPULATIONS
Tumors showing a T cell–inflamed tumor microenvironment also show evidence for NK cell presence (19). NKT cells and γδ TCR–expressing T cells have also been studied in this regard (20, 21).
Although both positive and negative immune regulatory functions have been attributed to these cell populations, the net effect is often positive, and it seems plausible that innate immune cells can contribute to tumor control either directly or indirectly, through DC activation or production of cytokines that support effector T cell differentiation.

A more recent paper from 2023 by Zhang et al. “Innate Immunity in Cancer Biology and Therapy”14 also describes how both innate and adaptive immunity work together to eliminate tumour cells:
In the innate immune system, myeloid-derived suppressor cells (MDSCs), macrophages, neutrophils, natural killer (NK) cells, dendritic cells (DCs), mast cells (MCs) and helper innate lymphoid cells (ILCs) have important regulatory effects on tumor progression [18].
Some innate immune cells have the capacity for detecting and eliminating tumor cells through various mechanisms, such as intrinsic cytotoxicity of NK cells and macrophage phagocytosis.
The interaction of the innate and adaptive immunity is exemplified by the uptake of tumor antigens by antigen-presenting cells (APCs), which results in cross-presentation and the priming of CD8+ T lymphocytes [19].
Innate immune cells are also involved in effector responses through antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cell cytotoxicity (ADCC) [20].

Another paper from 2023, “Exploiting innate immunity for cancer immunotherapy” by Yi et al.15 discusses how natural selective pressure causes immune-suppressive tumour cells to survive treatments and proliferate. Immune checkpoint inhibitors (ICIs) are the first line treatment for many cancers, but tumours may be resistant, or recurrences may occur.
Favouring innate immune mechanisms should improve the response to existing therapeutics and help to avoid resistance. Needless to say, as we have seen with mRNA/LNP products, any saRNA platform that inhibits innate immunity mechanisms is likely to be associated with treatment failures, recurrences and a worse prognosis:
Although these immunotherapies have achieved tremendous success in advanced cancers, some thorny issues remain to be resolved, including the unsatisfactory response rate and lack of accurate predictors.
It was estimated that 43.63% of all cancer patients were eligible for immune checkpoint blockade, and the overall response rate was below 13% in the US [43].
Besides, CAR-T cell therapy indications are limited to hematologic malignancies, without significant antitumor activity in solid tumors [44–47]. Generally, most clinically approved immunotherapies are T cell-centered. However, the effector functions of T cells are non-autonomous.
If your innate immunity has been compromised, then so is the adaptive immune response. This also applies to infections, not just tumour cells:
The initiation and sustainability of T cell response and the maintenance of T cell memory depend on innate immunity [48]. Innate immunity detects, captures, and processes cancer antigens and then triggers adaptive immunity.
At the same time, innate immune cells directly eradicate tumors by mounting their effector responses, such as the cytotoxicity of NK cells and the phagocytosis of macrophages [48].
Besides, due to the expression of Fc receptor (FcR) on macrophages and NK cells, innate immunity could participate in adaptive immunity by launching antibody-dependent cell cytotoxicity and phagocytosis (ADCC and ADCP) [49].
As the essential role of the innate immune arm in the onset, propagation, and maintenance of the cancer-immunity cycle, it is rationale to harness innate response to improve the current immunotherapy performance and relieve treatment resistance.
Forty to 60% of patients with advanced or metastatic melanoma respond to first-line immune checkpoint inhibitors (ICI) and responses are often durable [1]. However, half of all patients in the metastatic setting eventually progress. Retreatment or a therapy switch might improve tumor control among patients with advanced melanoma or in those with recurrence after adjuvant therapy.
From: “The concepts of rechallenge and retreatment with immune checkpoint blockade in melanoma patients” (2021)
https://www.sciencedirect.com/science/article/pii/S0959804921004275
Viruses, viral mimicry by HERVs, innate immunity, vaccines and disease
The prognosis for advanced brain cancers is very poor, with 10-year survival rates of between 14.9% for 0-14 year olds and 1.6% for 40+ year-olds.
Key takes from “Genetic secrets of long-term glioblastoma survivors” (2019) by Ivana Jovčevska16.
Glioblastomas are the most aggressive and lethal primary astrocytic tumors of the central nervous system. They account for 60% to 70% of all gliomas and the majority are diagnosed in Caucasian male patients at advanced age. Genetic analyses of glioblastoma show a great intra- and inter-tumor heterogeneity, which opens up a debate about its cellular origin.
Different types of brain cells, including astrocytes, neural stem cells, oligodendrocyte precursor cells and glioblastoma stem cells are proposed to have a role in tumor initiation and spreading; however, data is still inconclusive.
Two years is defined as “long-term survival” with glioblastoma:
Due to short life expectancy, long-term glioblastoma survivors are defined as patients who live longer than two years post-diagnosis. Extreme survivors, living 10 years or more after diagnosis, comprise less than 1% of all patients.
Glioblastoma used to be quite rare, but at Grade IV is the most aggressive type of glioma, and almost always rapidly fatal:
The World Health Organization (WHO) divides gliomas into four grades starting from pilocytic astrocytoma (Grade I), diffuse astrocytoma (Grade II), anaplastic astrocytoma (Grade III), and glioblastoma (Grade IV) [1,2]. Grades III and IV are considered high-grade gliomas and represent the majority of brain tumors [3].
Glioblastomas are astrocytic tumors with necrosis and microvascular proliferation. Patients suffering from this most malignant type usually succumb to the disease in 12 to 18 months after diagnosis [4].
Glioblastoma incidence is very low among all cancer types, i.e., 1 per 10 000 cases. However, with an incidence of 16% of all primary brain tumors it is the most common brain malignancy and is almost always lethal [5,6].
According to malignant progression, there are two types of glioblastomas: primary, originating de novo, and secondary, evolving from lower-grade gliomas.
What has this got to do with innate immunity? There is experimental data that demonstrated innate anti-tumour immune mechanisms resembling an antiviral response in glioblastoma, even without a virus being detected.
One may hypothesise that either prevention of cancer in the first place or an improved prognosis are a feature of having robust innate immunity. This may also be an indirect effect, due to the suppression of cancer-causing viruses such as Simian virus 40 (SV40) or human cytomegalovirus (CMV).
It would also explain why cases of cancers including hyperprogressive (turbo-) glioblastoma have significantly increased since the rollout of immune-suppressing experimental engineered mRNA products.
The authors appeared to be surprised that they were unable to confirm the presence of viruses as an explanation for most cases of IFN-associated innate immune responses and glioblastoma progression.
Key takes from “Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response” (2014) by Cosset et al.17:
In contrast to the studies reporting the presence of viral signatures in glioblastoma, no common or recurring active viruses were detected, despite finding an antiviral-like type I interferon response in some specimens. Our findings highlight a discrete and non-specific viral signature and uncharacterized short RNA sequences in glioblastoma.
Certain GBM specimens have been found to contain DNA sequences corresponding to the Simian virus 40 (SV40) large tumor antigen5,6 or human cytomegalovirus (CMV).7–11
CMV proteins have been suggested to promote tumor aggressiveness through increased tumorigenicity, invasion and angiogenesis.12
Type I interferon responses may be associated with poor survival for a subset of patients, but cases are inconclusive:
Furthermore, an antiviral response such as the upregulation of type I IFN/STAT1 genes correlates with poor survival outcome in a specific subtype of GBM patients.12 In line with these findings, anti-CMV treatment in GBM patients appears to extend survival rate.13
Yet contrary to these reports, other groups using similar methods did not detect CMV nucleic acids or proteins in GBM samples.14,15 Of note, such discrepancies have little correlation with the type of experimental methodology used in each of these studies. Investigations based on immunohistochemistry, polymerase chain reaction (PCR) or even short-term cultures on brain tumors lead to mixed results.7–9,11,14,15
The inconsistencies can be party attributed to the lack of positive infection controls (e.g., CMV positive glioma tissue) and the variable sensitivity of the end-point PCR used in these analyses.
Yet despite these confounding results, a recent consensus report argues there is sufficient evidence to conclude that CMV sequences and viral gene expression exist in most GBM.12
However, the same report highlights that: (i) to date, no study has demonstrated the production of infectious CMV virions by glioma and, (ii) the existence of CMV in glioma does not appear to fit classic definitions of active or latent infection, casting doubt on any direct influence on GBM.
They then discuss the mechanisms of innate immunity.
Note: Type I IFN responses may be induced in non-immune cells. This is a key feature of innate immune responses and a target for both mRNA and saRNA vaccines.
The initial stage of an antiviral response in non-immune cells involves induction of type I IFN and IFN-related genes including RnaseL, OAS, ISG15, PKR and Mx1.21 Therefore, we first characterized the type I IFN signaling response in the GBM specimens. These IFN-related genes (14 in total) were analyzed by qPCR in a collection of 33 biopsies from various tumors including GBM, tumors localized to the central nervous system, tumors in other peripheral organs and brain metastases from other primary cancer types.
Samples with elevated expression of IFN-related genes included three GBM, one squamous cell carcinoma, one invasive ductal carcinoma, one prostate adenocarcinoma, one breast carcinoma and one melanoma brain metastasis (Fig. 1; Supporting Information Table 1).
Some of the more significant genes affected:
ISG: Interferon-stimulated gene.
OAS: Oligoadenylate Synthetase, an interferon-induced antiviral gene.
MDA5: Anti-Melanoma Differentiation-Associated gene 5. Part of the RIG-I-like receptor family, which detect viruses.
IFITM3: interferon induced transmembrane protein 3. Another interferon-induced antiviral gene.
IRF4: Interferon regulatory factor 4, which functions as a key regulatory transcription factor in the development of immune cells, including T, B, plasma, dendritic cells and macrophages.
Dark blue on the GBM lines indicates a correlation with increased IFN-related gene expression:

Other GBM samples were distributed among subpopulations and exhibited low to moderate IFN-related gene expression levels. While we did indeed confirm increased gene expression levels in a substantial fraction of cancer biopsies (10/33), the expression patterns were not specific to GBM, and some biopsies did not express any IFN-associated genes (Fig. 1).
This could be an example of “survivorship bias”:

It is possible that most cases only progress to glioblastoma due to a lack of type I interferon responses:
Only a subpopulation of GBM samples exhibits an increase in expression of type I IFN-related genes, an observation supported by other reports.22 Accordingly, components of the IFN-related signaling pathway have previously been associated with acquired tumor radioresistance in a preclinical model of head and neck cell squamous cancer,23 and in breast, prostate and glioma cells.24
Some previous studies showed that IFN/STAT1 signaling controls antitumorigenic effects through upregulation of caspases,25–28 cyclin-dependent kinase inhibitor 1A (CDKN1A),29 the IFN-regulatory factor 1 (IRF1)/p53 pathway30 and downregulation of the Bcl2 (B-cell CLL/Lymphoma 2) family.31
Whatever is causing glioblastoma or the IFN-related innate immune response, they couldn’t pin it down to a known virus:
Traditional clinical diagnostics and HTS were subsequently used to exhaustively analyze the potential association between virus and GBM. Given the type I IFN gene expression profiles observed in some specimens, 20 GBM biopsies including the corresponding patient serum (upon availability), were screened via standard clinical diagnostic methods (semi-qPCR) for the presence of the following common neurotropic viruses: CMV, EBV, HSV, HHV6, MeV, PeV, JC virus, EV and VZV.
Although some biopsies were associated with a type I IFN-response (based on Mx1 and OAS1, 2 and 3 expression), none of the above-mentioned viruses were detected in any sample (Supporting Information Table 2).
Likewise, biopsies from three low-grade astrocytomas, one oligodendroglioma, two meningiomas, one ependymoma and one oligoastrocytoma were also found to be negative for these viruses.
Furthermore, brain metastases from tumors of a different histological origin (breast cancer and melanoma) also tested negative.
Only one meningioma biopsy gave a signal for EV at the LOD (Supporting Information Table 2).
CMV was not detected in any tumor, although some patients had an IgG positive serology, most likely from a past infection (absence of IgM).
This is why innate immunity is so critical for remaining cancer-free:
While no known viruses were found in any of the glioblastoma biopsies, the antiviral-like type I IFN gene activation signatures found in some samples could be explained by the fact that tumor infiltrating leukocytes produce IFNs, or by the sole effect of an antitumor immune response that produces IFNs.
Conclusion
This study is the first to harness HTS to comprehensively survey the virus landscape of GBM with corresponding experimental validation, and is designed to address the questions surrounding a possible virus–GBM association.
Previously, these questions were difficult to answer due to the inherent technological limitations of molecular analysis. Here we provide, for the first time, a complete answer to this question by developing a robust virus discovery pipeline with an accurate and elegant means of representation.
Moreover, we provide new insights reinforced by experimental validation and appropriate controls. We conclude that no known active human viruses, and notably no CMV signatures, were present in the GBM specimens analyzed, despite an antivirus-like, but non-specific, IFN response.
Altogether, our findings illustrate how a metagenomic HTS approach can provide a means of high-resolution virus screening and discovery in cancer, and highlight the urgency of reassessing the significance of ongoing clinical studies targeting active viruses in GBM.
The previous paper was from 2014, but I couldn’t find any reference to human endogenous retrovirus K (HERV-K).
However, a more recent paper from 2023 demonstrated the link between glioblastoma stem cells and HERV-K (= HML-2) activation.
Key takes from “Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma” by Shah et al.18:
Human endogenous retroviruses (HERVs) are ancestral viral relics that constitute nearly 8% of the human genome. Although normally silenced, the most recently integrated provirus HERV-K (HML-2) can be reactivated in certain cancers.
Here, we report pathological expression of HML-2 in malignant gliomas in both cerebrospinal fluid and tumor tissue that was associated with a cancer stem cell phenotype and poor outcomes.
Using single-cell RNA-Seq, we identified glioblastoma cellular populations with elevated HML-2 transcripts in neural progenitor–like cells (NPC-like) that drive cellular plasticity.
Using CRISPR interference, we demonstrate that HML-2 critically maintained glioblastoma stemness and tumorigenesis in both glioblastoma neurospheres and intracranial orthotopic murine models.
Additionally, we demonstrate that HML-2 critically regulated embryonic stem cell programs in NPC-derived astroglia and altered their 3D cellular morphology by activating the nuclear transcription factor OCT4, which binds to an HML-2–specific long-terminal repeat (LTR5Hs).
Moreover, we discovered that some glioblastoma cells formed immature retroviral virions, and inhibiting HML-2 expression with antiretroviral drugs reduced reverse transcriptase activity in the extracellular compartment, tumor viability, and pluripotency.
Our results suggest that HML-2 fundamentally contributes to the glioblastoma stem cell niche. Because persistence of glioblastoma stem cells is considered responsible for treatment resistance and recurrence, HML-2 may serve as a unique therapeutic target.
Naturally, the next step is to see if any papers investigated the link between HERVs and innate immune mechanisms. There is a link, but as ever it’s complicated.
In short, if you suppress HERVs with antivirals you may, paradoxically, be inhibiting evolutionarily long-established anti-tumour immune mechanisms.
HERV activation at the right time may have a role as a tumour suppressor via innate immunity mechanisms, yet it may promote progression of cancer and other diseases, at a later stage.
Key takes from “Endogenous Retroviruses as Modulators of Innate Immunity” by Russ & Lordanskiy19, also from 2023:
Although most HERVs are suspected to be 20 to 40 million years old, ERV integration into the human lineage has occurred for at least 100 million years [10,14,15].
The youngest HERVs are those belonging to the HERV-K (HML-2) subfamily, with integration events ranging between 200,000 and 35 million years ago [16,17,18].
Since the HML-2 subfamily is relatively young compared to other HERV clades, they spent the least amount of time accruing mutations. This has allowed the HML-2 subfamily to have the highest degree of coding competence, including multiple intact open reading frames for each viral gene (gag, pro, pol, and env) and over 60 proviral loci with full-length or near full-length sequences [8].
5. HERVs as Activators of the Innate Immune Response
Most PRRs in vertebrate biology can be groups into five major classes: (I) toll-like receptors (TLRs), (II) retinoic acid-inducible (RIG)-I-like receptors (RLRs), (III) C-type lectin receptors (CLRs), (IV) nucleotide oligomerization domain (NOD)-like receptors (NLRs), and V) absent in melanoma-2 (AIM2)-like receptors (ALRs) [70].
An important PRR which does not fall into these five major groups is cyclic GMP–AMP synthase (cGAS), due to its relatively unique mechanism of inducing downstream signaling (as discussed later) [71,72].
Perhaps there weren’t live viruses in the GBM patients, but there were HERVs:
While not all of these receptor classes have demonstrated the ability to recognize HERV-derived products, the viral-like nature of particular HERV-derived DNA, RNA, and proteins allows them to be recognized by and stimulate various PRRs to induce an innate immune response, similar to that of exogenous viruses.
“Aberrant HERV upregulation”, “viral mimicry” and disease:
As a result, the effects of aberrant HERV upregulation are said to mimic the anti-viral state that is induced following viral infection, leading to coinage of the term “viral mimicry” when discussing the implications of HERV re-activation on the innate immune response [37,73,74].
The authors then describe how different HERVs are found to be upregulated in patients with a range of diseases including multiple sclerosis (MS), rheumatoid arthritis (RA), lupus (SLE), and pulmonary arterial hypertension (PAH):
As previously discussed, MS patients typically express high levels of MSRV and other HERVs belonging to the HERV-W family. Specifically, there is a 1.5- to 3-fold increase in HERV-W Env expression, which acts as a TLR4:CD14 agonist and induces the expression of various pro-inflammatory cytokines to promote neuroinflammation [130].
While HERV-W Env antigen is detectable in the serum of 73% of MS patients (and 0% of healthy controls), it does not appear that HERV-W antigen levels are directly correlated to the severity of the symptoms [72]. However, from the same study, the HERV-W DNA copy number in PBMCs is increased in chronic progressive MS patients versus relapsing-remitting MS (RRMS) patients and healthy controls, suggesting that HERV-W proviruses undergo expression and re-integration events during active MS.
… HERV-K10 Gag1 protein shares an antigenic region with Collagen II, which is highly expressed in joints, and Freimanis et al. demonstrated that human fibroblast-like synoviocytes derived from a RA patient displayed several fold higher expression of HERV-K10 gag1 compared to samples from osteoarthritis (OA) and healthy donors [135]. The increase in HML-2 expression was confirmed by Reynier et al., who demonstrated that both types of HML-2 (HML-2 type I and type II) are significantly upregulated in the synovial fluid of RA patients compared to OA patients and healthy controls, with a higher HML-2 type I viral load associated with increased disease activity in RA patients [136].
Due to previous associations between HERV expression and SLE, Tokuyama et al. implemented RNA-sequencing on PBMCs derived from SLE patients and healthy controls to examine HERV expression on a locus-specific level using the software package they developed, ERVmap [141,142]. They identified over 100 unique ERV loci that are overexpressed in SLE patients and found that the total ERV read count significantly correlated with measures of disease severity, such as anti-nuclear, anti-double stranded DNA, anti-ribonucleoprotein (anti-RNP), and anti-Smith (Sm) anti-bodies. A separate study using the software package Telescope to characterize locus-specific HERV expression was released around the same time and confirmed these results, with over 300 loci significantly upregulated in SLE patients compared to controls in contrast to 10 downregulated loci [138].
Pulmonary arterial hypertension (PAH) is a progressive disorder that is characterized by endothelial dysfunction and vasculature remodeling of the pulmonary arteries, leading to obstruction of the blood flow and increased resistance [146,147]. The increase in blood vessel wall resistance causes an elevation in pulmonary artery pressure that can overload the right ventricular, resulting in heart failure and death.
To determine if lung tissue from PAH patients contained viruses that were previously implicated in PAH pathology (HIV, Human herpesvirus 8, and hepatitis C virus), Saito et al. performed a metagenomic viral screen using next-generation sequencing and did not detect the presence of any exogenous viruses [152].
… the authors observed a significant increase in HERV-K envelope and dUTPase mRNA in lung extracts from patients with PAH compared to healthy controls. Upon further inspection, they identified that HERV-K envelope and dUTPase proteins were primarily expressed in CD68+ macrophages that were present in the lung tissue and found that circulating monocytes from PAH patients exhibited higher levels of dUTPase mRNA compared to controls.
Interfering with innate immune mechanisms can have unforeseen effects years later that haven’t even been investigated in detail:
While it is difficult to definitively prove that HERV-K elevation is the (or an) initiating factor in PAH, the authors identified that HERV-K dUTPase is elevated in PAH patients, demonstrated the ability of HERV-K dUTPase to induce inflammation through the activation of several relevant cell types, and established that HERV-K dUTPase administration alone can result in pathological indicators of PAH development [152].
Though there is a relative lack of literature on the relationship between HERVs and cardiovascular-related diseases, it is clear that HERV-K elevation can instigate and sustain inflammation within the vasculature network.

In summary, you may think of HERVs as having an amplifying effect on both diseases and for helpful innate immune signalling. The older you get, the more pronounced the effects become. It’s one of the reasons why, when we get old, everything may seem to go wrong at once. An illness seems to trigger a cascade of other illnesses, seemingly out of proportion to the original condition.
Do mRNA “vaccines” upregulate HERVs? Whilst I was unable to find any papers investigating this directly, they are implicated by studies which found that SARS-CoV-2-induced inflammation and dysregulation of innate immunity contributed to the upregulation of many different HERVs.
Key takes from “Upregulation of Human Endogenous Retroviruses in Bronchoalveolar Lavage Fluid of COVID-19 Patients“ (2021) by Kitsou et al.20:
By comparing transcriptomes of bronchoalveolar lavage fluid (BALF) of COVID-19 patients and healthy controls, and peripheral blood monocytes (PBMCs) from patients and controls, we have shown that HERVs are intensely dysregulated in BALF of COVID-19 patients compared to those in BALF of healthy control patients but not in PBMCs.
In particular, upregulation in the expression of specific HERV families was detected in BALF samples of COVID-19 patients, with HERV-FRD being the most highly upregulated family among the families analyzed.
Multiple triggers, including mRNA “vaccines” and their proinflammatory cytokines, IgG imbalance, and suppression of innate immunity:
HERVs are remnants of ancient infections whose expression is upregulated in multiple conditions, including cancer and inflammation, and their expression is increased with increasing age.
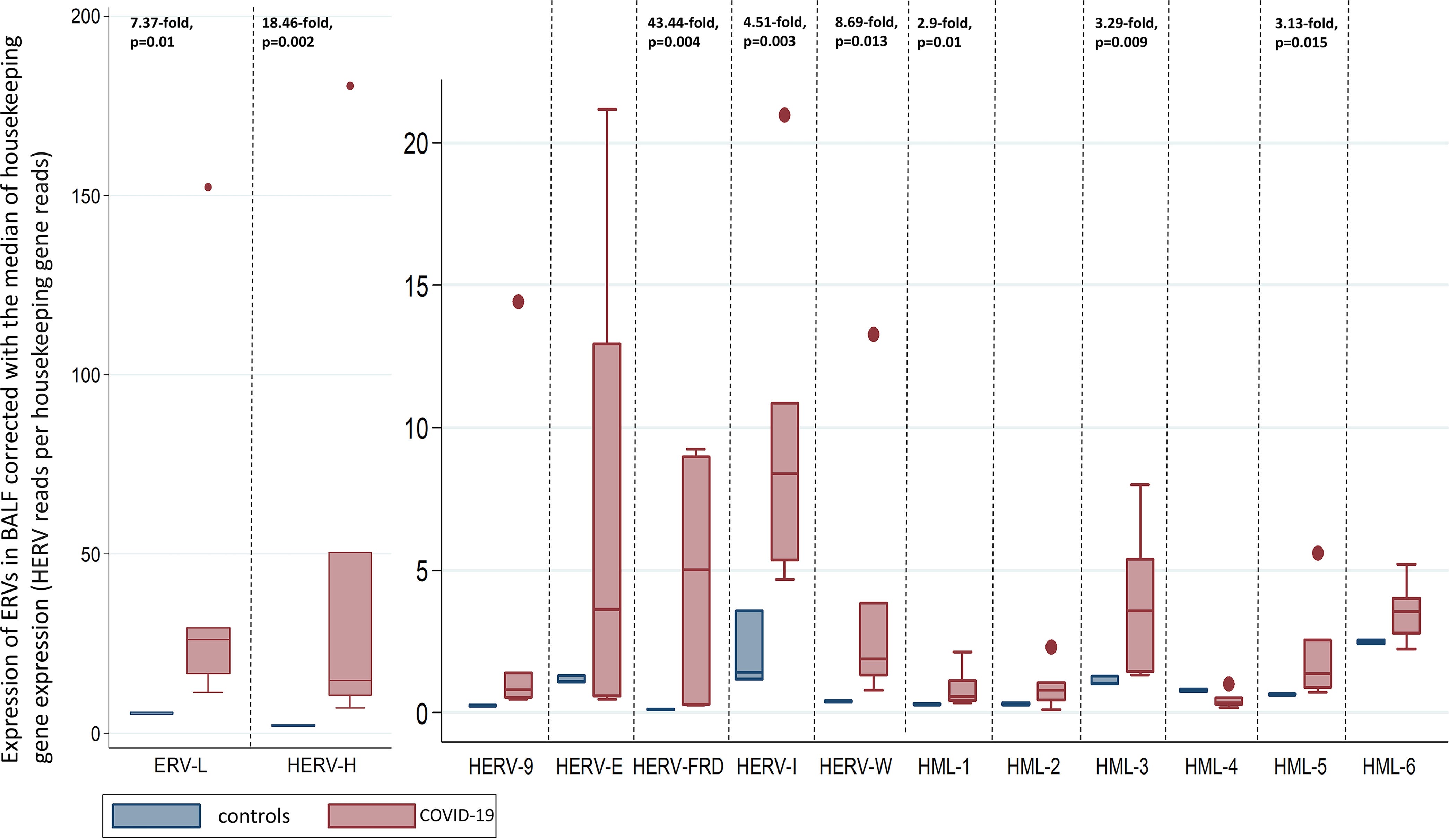
Our findings combined with data showing that entry factors for SARS-CoV-2 are coexpressed with innate immunity genes in respiratory cells suggest that HERV upregulation in BALF might indeed be relevant to local rather than systematic immunity responses (16).
A positive feedback mechanism, triggered by inflammation:
While HERVs are not commonly expressed throughout life, they have, on the other hand, been shown to have a dual role with respect to inflammation. They are upregulated by inflammatory pathways (17), but also ERV proteins and nucleic acids may trigger inflammatory responses (18).
For example, we have previously shown that IFI-16 has a broad range of single-stranded DNA (ssDNA) targets, and thus if HERVs or other transposable elements are upregulated and reverse transcribed as a result of COVID-19, this could amplify inflammatory responses (19).
Engineered mRNA “vaccines” mirroring viral mechanisms, again:
On the other hand, SARS-CoV-1 and SARS-CoV-2, alike, have been found to delay the production of interferons (20, 21), a mechanism that leads to reduced antiviral responses and hence has been speculated to promote the cytokine storm that has been described in severe COVID-19.
In some patients developing severe COVID-19, this immune dysregulation has been linked to the presence of IgG auto-antibodies against type I INFs that preexisted before the infection and to inborn errors in the regulation of type I IFN innate immunity (22, 23).
Don’t interfere with things that you don’t understand. One outcome may be cytokine storm, another is multisystem inflammatory syndrome (MIS)… or cancer and other diseases discussed previously:
It is reasonable to assume that the intricate mechanisms behind the inflammatory responses to the SARS-CoV-2 infection and its development to severe COVID-19 with cytokine storm in patients prone to severe illness are the result of the aberrant balance in the transcription of proinflammatory, anti-inflammatory, and antiviral immune cytokines, which may be highly influenced by the effect of exogenous and endogenous viral pressure on innate immunity.
Concluding remarks
Although the manufacturer may choose to use these patented IIPs there is a viral protein group that also acts as an IIP that has to be used. Without it, a replicon vaccine is no longer a replicon vaccine.
Viral evolution has given several roles to non-structural replicase proteins (nsPs 1, 2, 3 and 4). One of these is to be involved in the replication of the input genomic RNA, hence the name, and why it is an essential step.
The other role is to suppress innate immune mechanisms, which is necessary for both viral survival and for replicon vaccine function.
The Japanese recipients of Kostaive are discovering the consequences of this.

Part 4 will continue the discussion, with a focus on the patent landscape and the effects of dsRNA contamination.


References
Chen, Xiaohua, Taotao Zhang, Wei Su, Zhihui Dou, Dapeng Zhao, Xiaodong Jin, Huiwen Lei, et al. ‘Mutant P53 in Cancer: From Molecular Mechanism to Therapeutic Modulation’. Cell Death & Disease 13, no. 11 (18 November 2022): 1–14. https://doi.org/10.1038/s41419-022-05408-1.
Yoon, Cheol-Hee, Eun-Soo Lee, Dae-Seog Lim, and Yong-Soo Bae. ‘PKR, a P53 Target Gene, Plays a Crucial Role in the Tumor-Suppressor Function of P53’. Proceedings of the National Academy of Sciences 106, no. 19 (12 May 2009): 7852–57. https://doi.org/10.1073/pnas.0812148106.
PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice | Blood | American Society of Hematology. Accessed January 31, 2025. https://ashpublications.org/blood/article/126/13/1585/105435/PKR-inhibits-the-DNA-damage-response-and-is
Watanabe T, Imamura T, Hiasa Y. Roles of protein kinase R in cancer: Potential as a therapeutic target. Cancer Sci. 2018;109(4):919-925. doi:10.1111/cas.13551
Beattie, E., J. Tartaglia, and E. Paoletti. ‘Vaccinia Virus-Encoded eIF-2α Homolog Abrogates the Antiviral Effect of Interferon’. Virology 183, no. 1 (1 July 1991): 419–22. https://doi.org/10.1016/0042-6822(91)90158-8.
Sharp, T. V., J. E. Witzel, and R. Jagus. ‘Homologous Regions of the Alpha Subunit of Eukaryotic Translational Initiation Factor 2 (eIF2alpha) and the Vaccinia Virus K3L Gene Product Interact with the Same Domain within the dsRNA-Activated Protein Kinase (PKR)’. European Journal of Biochemistry 250, no. 1 (15 November 1997): 85–91. https://doi.org/10.1111/j.1432-1033.1997.00085.x.
Guo, Liang, Yayun Chi, Jingyan Xue, Linxiaoxi Ma, Zhiming Shao, and Jiong Wu. ‘Phosphorylated eIF2α Predicts Disease-Free Survival in Triple-Negative Breast Cancer Patients’. Scientific Reports 7, no. 1 (15 March 2017): 44674. https://doi.org/10.1038/srep44674.
Ramelot, Theresa A., John R. Cort, Adelinda A. Yee, Furong Liu, Michael B. Goshe, Aled M. Edwards, Richard D. Smith, Cheryl H. Arrowsmith, Thomas E. Dever, and Michael A. Kennedy. ‘Myxoma Virus Immunomodulatory Protein M156R Is a Structural Mimic of Eukaryotic Translation Initiation Factor eIF2α’. Journal of Molecular Biology 322, no. 5 (4 October 2002): 943–54. https://doi.org/10.1016/S0022-2836(02)00858-6.
Mattoo S ul S, Kim SJ, Ahn DG, Myoung J. Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses. 2022;14(3):530. doi:10.3390/v14030530
Jin R, Cao X, Lu M, Gao Q, Ma T. The intersection molecule MDA5 in Cancer and COVID-19. Front Immunol. 2022;13:963051. doi:10.3389/fimmu.2022.963051
orf4 - Non-structural protein 4 - Human coronavirus HKU1 (HCoV-HKU1) | UniProtKB | UniProt. Accessed January 31, 2025. https://www.uniprot.org/uniprotkb/Q0ZJ92/entry
Krajewska M, Moss SF, Krajewski S, Song K, Holt PR, Reed JC. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res. 1996;56(10):2422-2427.
Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445-474. doi:10.1146/annurev-immunol-032414-112043
Zhang Y, Xue W, Xu C, et al. Innate Immunity in Cancer Biology and Therapy. Int J Mol Sci. 2023;24(14):11233. doi:10.3390/ijms241411233
Yi M, Li T, Niu M, et al. Exploiting innate immunity for cancer immunotherapy. Mol Cancer. 2023;22(1):187. doi:10.1186/s12943-023-01885-w
Jovčevska I. Genetic secrets of long-term glioblastoma survivors. Bosn J Basic Med Sci. 2019;19(2):116-124. doi:10.17305/bjbms.2018.3717
Cosset É, Petty TJ, Dutoit V, et al. Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response. Int J Cancer. 2014;135(6):1381-1389. doi:10.1002/ijc.28670
Shah AH, Rivas SR, Doucet-O’Hare TT, et al. Human endogenous retrovirus K contributes to a stem cell niche in glioblastoma. J Clin Invest. 133(13):e167929. doi:10.1172/JCI167929
Russ E, Iordanskiy S. Endogenous Retroviruses as Modulators of Innate Immunity. Pathogens. 2023;12(2):162. doi:10.3390/pathogens12020162
Kitsou K, Kotanidou A, Paraskevis D, et al. Upregulation of Human Endogenous Retroviruses in Bronchoalveolar Lavage Fluid of COVID-19 Patients. Microbiology Spectrum. Published online October 6, 2021. doi:10.1128/Spectrum.01260-21
Cyprinus Carpio - Part 3 of another magnum opus.
From Part 1: "The active-control vaccine (Comirnaty) used in the Japanese phase III study (Study ARCT-154-J01) is a replicon-non-containing RNA vaccine."
A VRP by another name. Not unrelated...
From Part 3: "Although the manufacturer may choose to use these patented IIPs there is a viral protein group that also acts as an IIP that has to be used. Without it, a replicon vaccine is no longer a replicon vaccine."
It's the mimicry that gets one to hold their beer, and it works something like this... https://youtu.be/HrENha7E1xc?
Can't wait for Part 4. FYI - Your up on Ah Kahn Syed's latest.
All of these extremely complex immune pathways doing different things at different times for different reasons causing different effects for different people should never have been interfered with.It was and is madness, the mRNA platform should never have left the theoretical realm. The immune system is so complex we had no business experimenting with it on humans. Our immune system is the best defence barrier we have against many diseases, to interfere with that complex structure is unconscionable, that is what has been done on a worldwide scale with no true understanding of the possible outcomes.