


SARS-CoV-2, spike protein, mitochondrial hijacking & associated diseases

Reading time:
short story - novelette - novella - novel - PhD thesis - War and Peace - U.S. Tax Code
TL;DR: However bad you may have thought it was, its probably worse.
Updated:
26th December ‘23: (Melatonin: a potent protector of mitochondria).
Also available in:
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.

Contents
Mitochondrial hijacking by cancer cells
SARS-CoV-2 and mitochondrial disfunction
SARS-CoV-2 and mitochondrial gene down-regulation
Spike protein impairment of cardiac mitochondria
Background
Some time in 2022 I was sent a screenshot of part of paper discussing transfection of mitochondria by mRNA “vaccines”, mtDNA integration and expression. Quite explosive stuff I think you will agree, and I started reading around the subject.
To write about it I needed the full paper. Some time later I managed to get the software to do an OCR conversion, extracted enough text for a search and finally struck gold.
Its called “Functionality and Clinical Effects of Anti-Cov2 Vaccines (Aka Mrna) And Integration on Mitochondrial DNA”, was written by Dr. Raffaele Ansovini, and published in 2022 in “World Journal of Clinical & Medical Images (WJCMI)”.
Last week I started critiquing the paper, and things quickly started to fall apart.
WJCMI peer review their submissions. This process usually takes 2-3 months including rewrites, and your paper may be rejected even then (censorship aside), so the first red flag was the 1 week turnaround:

There is a hit piece on so called “predatory journals” here:
https://predatoryreports.org/news/f/list-of-all-opast-publishing-group-predatory-journals
Without going through it line by line the following statement demanded references, to put it mildly:
5. The vaccine mRNA does not refer to the segment relating to the Spike protein, but to the entire viral genome.
In fact it was a full stop. No point in reading on because unless you can substantiate such a remark everything else is called into question. It should have been sent back on this point alone.
And it gets better:
6. Therefore, the Ribosomes are not the final destination of the vaccine mRNA; instead, the Mitochondria are actually the seat of a DNA, accommodating the SARS-Cov2 genome in the same way as it does in the nuclear genome.
18. f) DNA Helicase which by copying the inserted DNA - DNA from the mRNA vaccine - makes it more permanently stored in the mitochondrial genome.
19. - Note - The presence of DNA Helicase in mRNA vaccines is further proof that integration does not take place in the core genome. It is impossible for the nuclear genome to simultaneously absorb and copy a viral genome when, due to the 'restitutio ad integrum' acquired during evolutionary times, it tends to shed the viral genome after the infection is past. If this were not the case, the 'Spanish flu' contracted by our grandparents could reappear today through genetics.
21. A larval infection of low-intensity SARS Cov2 has thus occurred at the mitochondrial site.
22. This infection is evidenced by the presence of biochemical patterns associated with the existence of a viral cycle in the blood of multi-vaccinated with SARS vaccine- Cov2.
No experiments were conducted so I was eager to see which work could be used to support these hypotheses. Unfortunately this crock was full of sauce but no beans:

In case you are wondering I did follow up a couple of these, but soon lost the will to live.

He supposedly did a BLASTP search for enzyme and protein matches:

I’m not dismissing everything he says by any means, but without independent references I just cannot use it. Line-by-line interpretation is very time consuming both for myself and for you, the inquisitive reader, and I won’t waste any more time on it.
But by all means take a peak for yourself:
I did say that I read around the subject and the irony here is that if he’d included them there are plenty of jaw-dropping research papers he could have cited. This Substack should be the paper that his could have been.
Introduction
We need to start by defining in simple terms what mitochondria are and what there main function is:
Mitochondria are membrane-bound cell organelles (mitochondrion, singular) that generate most of the chemical energy needed to power the cell's biochemical reactions. Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother.

From: “MITOCHONDRIA“ (2023)
The main role performed by mitochondria is to generate ATP. Adenosine triphosphate (ATP) is the source of energy for use and storage at the cellular level. Large amounts must be generated each second by mitochondria as it cannot be stored. Cells contain from 1000 to 2500 mitochondria and this function is so important that they can take up as much as 25% of a cells volume.
Each ATP needs to be recycled from ADP 1000 times to meet the average cells requirement of 10 billion ATP per day. In any given instant we have around 250 g of ATP in our cells, with an energy equivalent of 4.25 Watts, or 1.2KW per day in a healthy person. The brain uses around 70% of this, which helps explain why there is a strong correlation between mitochondrial dysfunction and neurodegeneration.1
Mitochondrial hijacking by cancer cells
Whilst researching this, the kind of hijacking I expected was perhaps increased expression of mitochondrial DNA-encoded genes or maybe suppression of mitochondria in cytotoxic T-cells. And whilst there is plenty of evidence for this23 an in vitro study published in 2021 by Saha et al4 revealed a more macabre reality - hijacking in the more literal sense.
Baldwin & Gattinoni sets the scene (emphasis mine):5
The Thirty-Six Stratagems is a collection of warfare wisdom from ancient China that has been distilled over the centuries into short proverbs that illustrate how to implement cunning and deception in war and politics. One of the key stratagems in order to outwit opponents on the battlefield is 釜底抽薪 (to ‘Steal the firewood from under the pot’). Rather than attacking the enemy’s fighting forces, the essence of this tactic is to attack their ability to wage war by taking the fuel out of the fire. As often is the case, life imitates art. Now, writing in Nature Nanotechnology, Saha et al.1 provide evidence that cancer cells outwit the ability of immune cells to wage war on them by stealing their mitochondria, that is, the firewood that fuels the cells.
From: “Cancer cells hijack T-cell mitochondria“ (2021)
I won’t go into great depth here, but its worth adding that this is possibly another reason why cancer vaccines always seem to over-promise and under-deliver, antigenic shifting by the tumour being the principle reason. Cancer cells can be very sneaky - natural selection makes them this way, only the most evasive cells can survive to proliferate:
“Thus, there is an urgent need to develop vaccines against neoantigens to convert immune “cold” tumors to “hot” ones. Nevertheless, the development of novel antigens is complex. Tumor growth is an evolutionary process, and the genomes of tumor cells are constantly mutating as they grow. Vaccines developed for the genome of a single tumor sample often have little success when the neoantigen is a subclone of a mutated tumor gene. 79 Furthermore, the variation in neoantigens produced by tumors in different patients makes treatment against neoantigens extremely challenging.84,85
From: “Igniting Hope for Tumor Immunotherapy: Promoting the “Hot and Cold” Tumor Transition“ (2022)
Cells can communicate with other cells by connecting via nanotubes. Amyloid-β6, SARS-CoV-27, HIV8 and many other pathogens also uses these to infect neighbouring cells.
In hijacking mitochondria via nanotubes the defending cancer cell grows stronger at the expense of the attacking cytotoxic T-cell.

PD1 = Programmed cell death protein 1. PD1 is a checkpoint receptor on the surface of T and B cells. Its effect is to down-regulate the immune system and promote self-tolerance. This is great for preventing autoimmune diseases, but its also used by many cancer cells to prevent cytotoxic T cell inflammatory responses.
α-PD-1 mediated immune checkpoint blockade is used as a first line therapy for melanoma and lung cancer, and a second line treatment for squamous cell carcinoma of the head and neck (SCC-HN), and other tumour types.9 Mitochondrial hijacking also limits the effectiveness of this sort of therapy.
The researchers found that nanotube inhibitors prevented hijacking and restored the action of checkpoint inhibitors. At first it may sound like an effective treatment, but nanotubes facilitate so many essential trafficking functions that total inhibition would quickly lead to all manner of disorders.
Indeed a study using the same nanotube inhibitor from the study (dual farnesyltransferase and geranyl-geranyl transferase inhibitor L-778,123) led to dose limiting outcomes for 2 of the 4 patients on the higher dose of a trial which aimed to increase radiosensitivity of advanced pancreatic cancer.
Grade 3 events are serious and interfere with a person's ability to do basic things like eating or getting dressed. Grade 3 events may also require medical intervention, (emphasis mine):
Results: There were no dose-limiting toxicities observed in the eight patients treated on dose level 1. Two of the four patients on dose level 2 experienced dose-limiting toxicities consisting of grade 3 diarrhea in one case and grade 3 gastrointestinal hemorrhage associated with grade 3 thrombocytopenia and neutropenia in the other case. Other common toxicities were mild neutropenia, dehydration, hyperglycemia, and nausea/vomiting. One patient on dose level 1 showed a partial response of 6 months in duration.
From: “A Phase I Trial of the Dual Farnesyltransferase and Geranylgeranyltransferase Inhibitor L-778,123 and Radiotherapy for Locally Advanced Pancreatic Cancer“ (2004)
Another trial used L-778,123 and was even less successful.
Grade 4’s are life threatening or disabling adverse events and require immediate medical attention. QT(c) interval prolongation is an irregularity of the electrical activity of the heart that places patients at risk of ventricular arrhythmias:
Twenty-five patients received 51 complete courses of L-778,123. An unacceptably high incidence of dose-limiting toxicities, consisting of grade 4 thrombocytopenia, significant prolongation of the QT(c) interval, and profound fatigue, was observed at the 1120 mg/m(2)/day dose level.
From: “A phase I and pharmacological study of the farnesyl protein transferase inhibitor L-778,123 in patients with solid malignancies“ (2001)
Graphical abstract from the Saha et al paper, illustrating mitochondrial hijacking:

Transfection of mitochondria
Although Dr. Ansovini didn’t cite any studies, there has actually been a significant amount of research into this.
Quick takes from a few PubMed studies:
“Transfection of mitochondria: strategy towards a gene therapy of mitochondrial DNA diseases”10 (1995) was an in vitro study into how transfection of mitochondria could be used to replace a mutant mitochondrial genome.
“Targeting repair proteins to the mitochondria of mammalian cells through stable transfection, transient transfection, viral transduction, and TAT-mediated protein transduction”11 (2009) described how plasmids or protein transduction domains could be developed to stably transfect cell lines in order to express a mitochondrial-targeted chemotherapeutic protein of interest.
“Transfection of miR-31* boosts oxidative phosphorylation metabolism in the mitochondria and enhances recombinant protein production in Chinese hamster ovary cells”12 (2021) is, from what we know now13, a disturbing study into how the mature miRNA mimic sequence for microRNA miR-31 (5′-UGCUAUGCCAACAUAUUGCCAUC-3′) could be transfected into Chinese hamster ovary cells (CHO’s).
The objective was to reprogram mitochondrial energetic metabolism in order to increase the production of therapeutic recombinant proteins, such as monoclonal antibodies. If the mitochondria have all this spare respiratory capacity why not supercharge them and decrease the inefficient Warburg effect of the cell?
If they wanted to, ahem, transfect oncogenic miR-2114 the sequence is UAGCUUAUCAGACUGAUGUUGA, and we have sequences for both primary and precursor miRNA constructs for reference.15
The process to derive homologous miRNA from synthetic mRNA is as follows, and one of the ways that miR-21 promotes cancer is by inhibiting mitochondrial-mediated apoptosis.16

From “MicroRNA therapeutics: towards a new era for the management of cancer and other diseases”, “Overview of microRNA (miRNA) biogenesis”, source: https://www.nature.com/articles/nrd.2016.246 (paywalled). “The TFAMoplex—Conversion of the Mitochondrial Transcription Factor A into a DNA Transfection Agent”17 (2022) used repurposed human protein mitochondrial transcription factor A (TFAM) nanoparticles in an in vitro study to create a “safe and efficient” DNA transfection agent. Endogenous TFAM is expressed in the cytoplasm and transported to the mitochondria, where it regulates the mitochondrial genome.

From “Figure 7. DNA uptake by HeLa cells.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8922101/ “Nanoparticles for vaccine and gene therapy: Overcoming the barriers to nucleic acid delivery“18 (2022) includes a section on how “a third target location for nucleic acid therapeutics is the mitochondria”. They discuss how mitochondria have their own genome with a limited capability to synthesise mitochondrial proteins, although most are expressed in the cytoplasm. Mutations in mitochondrial DNA (mtDNA) can cause several inherited diseases and nucleic acid gene therapies using RNAs or DNAs could be used to either replace or edit these and prevent the synthesis of abnormal proteins.
The authors note that just because you have delivered a nanoparticle to a cell doesn’t mean that it will also be successfully delivered to the nucleus or mitochondria. This challenge can be addressed by using a leader sequence from mtDNA in the nucleic acid, as this can be recognised by mitochondrial membrane translocators.
In relation to this, in 2021’s “Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines”, Xia ran the sequences for Pfizer’s BNT162b2 and Modernas mRNA-1273 against human genome databases.19 He matched a 139 nt segment against 959 nt long mitochondrially encoded 12S ribosomal RNA (=”12S” or “12S rRNA” or “mtRNR1”).
It is very telling from the author why they took this approach. By including mitochondrial RNA ribosomal expression could quickly be optimised:
SELEX: “Systematic evolution of ligands by exponential enrichment” or “in vitro selection” or “in vitro evolution” is used for isolating high-affinity single-stranded (ss) DNAs or RNAs from a large library with random sequences:20
“I have previously mentioned different approaches for optimizing 5′-UTR and 3′-UTR. Given sufficient time, the systematic evolution of ligands by exponential enrichment (SELEX) [54] should be the preferred method. However, in an emergency, the alternative approach of borrowing from nature could be more efficient.”
“The design of the 3′-UTR of the Pfizer/BioNTech mRNA vaccine is a combination of SELEX and borrowing from nature. The objective is to find naturally occurring RNA segments that perform better than the 3′-UTR of human β-globin mRNA [54]. Two RNA segments outperform other alternatives through the SELEX optimization protocol [54]. One of them is from the human mitochondrial 12S rRNA (mtRNR1), and the other segment is from human AES/TLE5 gene.”
Mutations in this gene are associated with sensorial hearing loss and the rRNA it encodes regulates insulin sensitivity and metabolic homeostasis. Deficiencies in downstream cytochrome c oxidase affect the mitochondrial respiratory chain, resulting in cardiomyopathy, encephalomyopathy, weak muscles or liver damage, amongst others.21

“Changes that occur in mitochondria due to aging or aging related disorders are associated with a reduction in the function of mitochondria.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9570330/ If there are autoimmune antibodies to 12S then these are the pathologies to be aware of, and this is quite possible due to overexpression and extramitochondrial accumulation of the epitopes. Cytosolic mtRNA is recognised by retinoic acid-inducible gene I (RIG-I) and can trigger RIG-I-MAVS-dependent innate autoimmune responses, including Lupus and amyotrophic lateral sclerosis (ALS).22
Naturally the next step is a literature search to see if this expedited “borrowing from nature” of a 12S segment may have real world consequences. And a few papers are quite revealing. I could easily fill a few Substacks just reviewing 12S:
MOTS-c (mitochondrial ORF of the 12S rRNA type-c) “is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis”. MOTS-c expression is associated with longer lifespans and intermittent treatment improves the physical capacity of older mice.23
When we are stressed MOTS-c, a mitochondria-derived peptide (MDP), can translocate to the nucleus and affect transcriptional responses to stress, which is a characteristic of a health and lifespan extending mitokine.24
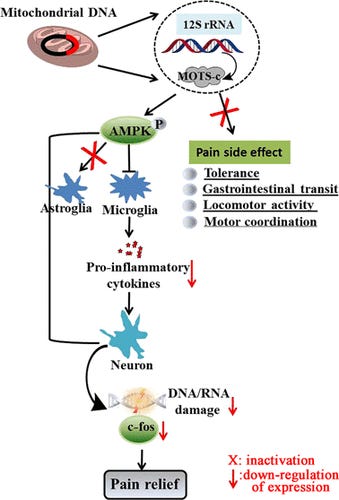
Returning to insulin sensitivity, a study using mice found that MOTS-c treatment can prevent T-cell mediated destruction of pancreatic islets, preventing type 1 diabetes (T1D) via the TCR/mTORC1 pathway, preventing a shift to the autoimmune phenotype.25 Apart from preventing certain autoimmune disorders, MOTS-c can functionally prevent metabolic disorders26 and reduce neuropathic pain by inhibiting activation of components of the neuroimmune system (microglia) and preventing neuronal oxidative damage.27

MOTS-c can ameliorate neuropathic pain. Source: https://pubs.acs.org/doi/full/10.1021/acschemneuro.3c00140 
“Physiological significance of the MOTS-c protein. MOTS-c is encoded from a region within the 12S rRNA MT-RNR1 gene. The MOTS-c protein has both inhibitory effects (inflammation, age-related disorder, apoptosis) and also promotes healthy functioning in brain and other tissues. bp, base pair.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9570330/ Tying in nicely to the last Substack on MS,28 a clinical trial involving 43 patients with relapsing-remitting MS and 41 healthy controls found that the MS patients had significantly lower MOTS-c values, and this may help explain their higher incidence of diabetes, obesity, higher blood lipids and cardiovascular disease. The study from 202229 discusses how chronic inflammation associated with MS mediates the production of reactive oxygen species (ROS).
ROS can then induce mitochondrial DNA mutations, eventually damaging the mitochondrion and inducing metabolic stress with protein misfolding in the cell’s endoplasmic reticulum (ER).
If this happens in the CNS then disrupted energy production can cause damage to neurons and finally to the whole nerve axon. You will have realised by now this is a positive feedback mechanism as the damaged neurons further trigger microglia in an autoimmune response.
Also be aware that MOTS-c autoantibodies aren’t required to kick the cycle off, vax induced ROS would suffice. That’s the theory, but is it happening in vivo? Yes, in a word, and I review some studies later.
Supplementation with MOTS-c
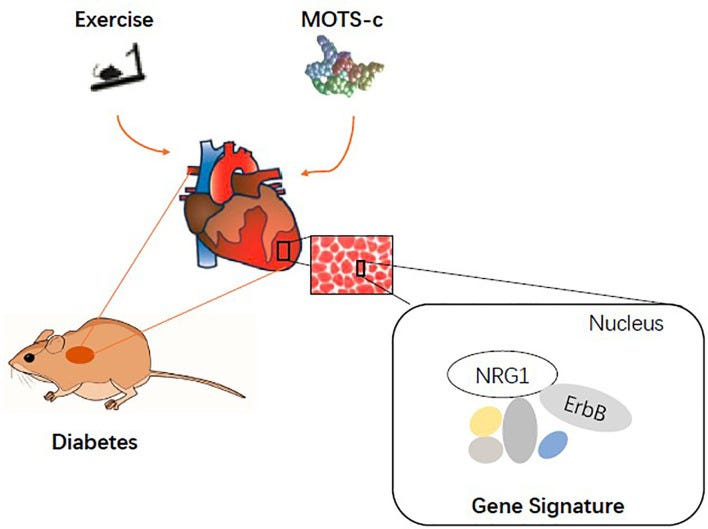
For those of us that prefer sofa surfing to the real thing (hat tip to Jessica Rose) you will be pleased to learn that a study using rats from 2022 concluded that MOTS-c supplementation mimicked exercise-induced cardio-protection against artificially induced diabetes. Both methods activated the protective neuregulin1 (NRG1) to ErbB4 signalling pathway.
“ErbB is a receptor for NRG1, a cardiomyocyte mitogen that increases myocardial angiogenesis in rats with diabetic cardiomyopathy (42). The NRG1/ErbB pathway also induces cardiomyocyte proliferation and regulates angiogenesis and vascular endothelial function (43–45).”30

“Model depicting the overlapping mechanisms between exercise and MOTS-c supplements in restoring cardiac function in diabetes.” source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8969227/ However I would always recommend changing lifestyle, including diet and exercise before you supplement, or to complement it. And the price of human MOTS-c peptide may put you off. A more economical way would be to limit mitochondrial damage in the first place by taking antioxidants, many of which also promote recovery of mitochondria too.
These include baicalin, turmeric, CBD, quercetin, berberine, vitamin D, NAC,31 resveratrol, and epigallocatechin-3-gallate.32
An exception to this in favour of supplementation is where exercise may be contraindicated, such as after suspected myocarditis or with ME/CFS.33

“Natural products trigger mitochondrial biogenesis. Natural products such as berberine, resveratrol, and epigallocatechin-3-gallate trigger mitochondrial biogenesis through regulating the AMPK-SIRT1-PGC-1α pathway.” Source: https://www.frontiersin.org/articles/10.3389/fphar.2023.1093038/




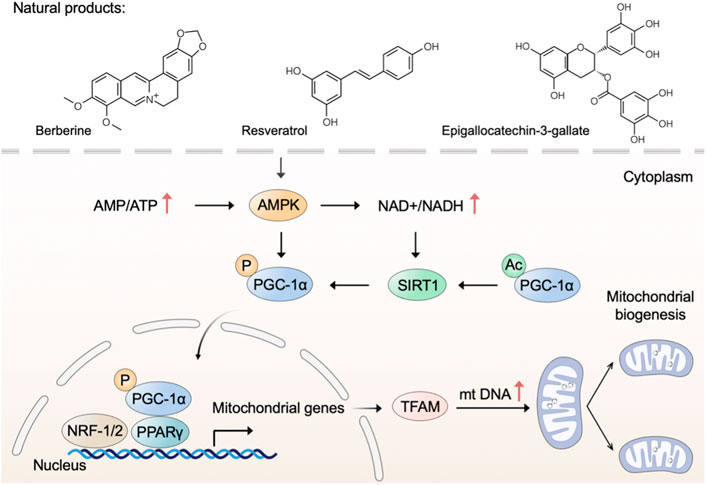
One of my favourite therapeutics, berberine also possesses “magic bullet” properties that pharma can only envy. Reading the literature you see that those that create can also destroy, and one of the ways that berberine and other therapeutics induce cancer cell apoptosis is by mitochondrial‑mediated apoptosis.34
It also selectively accumulates in cancer cell mitochondria, inhibiting growth of the tumour by multiple mechanisms as well as working synergistically with some chemotherapy drugs.35
Added 26th December ‘23:
Melatonin: a potent protector of mitochondria
Due to the risk of population level increases in all conditions linked to long term mitochondrial dysfunction, particularly cardiomyopathy and diabetes, its important to highlight the best of the best options that are both accessible and affordable by most people.
Thanks to Just a Clinician for suggesting melatonin and this link:
This paper would seem to ft here: Melatonin as an antioxidant: under promises but over delivers https://onlinelibrary.wiley.com/doi/10.1111/jpi.12360
It could easily justify a dedicated future Substack.
And thanks to my colleague Dr Alberto Rubio-Casillas for the second link. Both were co-authored by Dr. Russell J. Reiter:
Regarding spike-induced damage to the mitochondria, we have a good ally: melatonin.
This article was written by Dr. Russell Reiter, the leading expert on melatonin.
Melatonin is a popular therapeutic with relatively low toxicity. Its main market is for the treatment of sleep disorders. I can link to a post describing dosing strategies, side effects and QC issues but cannot make any personal recommendations as we all have different tolerances and medical & drug histories.36
Perhaps not unconnected to the previous recommendation, Scutellaria baicalensis is also a source of phytomelatonin that may reach a pharmacologically active dose when baicalin is taken as recommended. I discussed it here and in the linked OSF submission:

Coffee is another source, although again we are only talking micrograms per cup:
...To date, the phytomelatonin content of coffee beans is the highest recorded in plant material. Roasted coffee beans showed an even higher phytomelatonin content than green beans, reaching 60–78 ng of phytomelatonin per mL of brewed coffee [34].
From: “The Potential of Phytomelatonin as a Nutraceutical” (2018)
Key takes in relation to mitochondria from “Melatonin as an antioxidant: under promises but over delivers“37 (2016, emphasis mine):
Melatonin is uncommonly effective in reducing oxidative stress under a remarkably large number of circumstances. It achieves this action via a variety of means: direct detoxification of reactive oxygen and reactive nitrogen species and indirectly by stimulating antioxidant enzymes while suppressing the activity of pro-oxidant enzymes.
…melatonin also reportedly chelates transition metals, which are involved in the Fenton/Haber–Weiss reactions; in doing so, melatonin reduces the formation of the devastatingly toxic hydroxyl radical resulting in the reduction of oxidative stress.
Melatonin's ubiquitous but unequal intracellular distribution, including its high concentrations in mitochondria, likely aid in its capacity to resist oxidative stress and cellular apoptosis. There is credible evidence to suggest that melatonin should be classified as a mitochondria-targeted antioxidant.
Experimental findings also indicate that melatonin renders treatment-resistant cancers sensitive to various therapeutic agents and may be useful, due to its multiple antioxidant actions, in especially delaying and perhaps treating a variety of age-related diseases and dehumanizing conditions.
Melatonin has been effectively used to combat oxidative stress, inflammation and cellular apoptosis and to restore tissue function in a number of human trials; its efficacy supports its more extensive use in a wider variety of human studies. The uncommonly high-safety profile of melatonin also bolsters this conclusion.
…melatonin reportedly limits electron leakage from the mitochondrial respiratory chain leading to fewer oxygen molecules being reduced to the superoxide anion radical; this process is referred to as radical avoidance.
…a major theory of aging, that is, the mitochondrial theory, incriminates damage to these organelles as being responsible for the aging of cells, of organs and of organisms.159-161 As oxidative damage of mitochondria is central to a number of serious pathologies and to aging, conventional antioxidants should be useful in forestalling these diseases or delaying degenerative processes associated with advanced age.
One reason for the failure of conventional antioxidants to ameliorate the severity of ROS-related diseases may be a result of their inability to concentrate in mitochondria where free radical production is maximal. Thus, it seemed like a worthwhile strategy would be to develop mitochondria-targeted antioxidants; this has been done and they were shown effective in reducing mitochondrial damage and the resulting apoptosis.
As an example…triphenyl phosphonium cation (TPP).170 Combining TPP with Q10 allowed the resulting molecule, called MitoQ, to rapidly cross the cell and mitochondrial membranes and to accumulate in concentrations up to several hundred-fold greater in the mitochon
drial matrix than that of the unconjugated antioxidant.
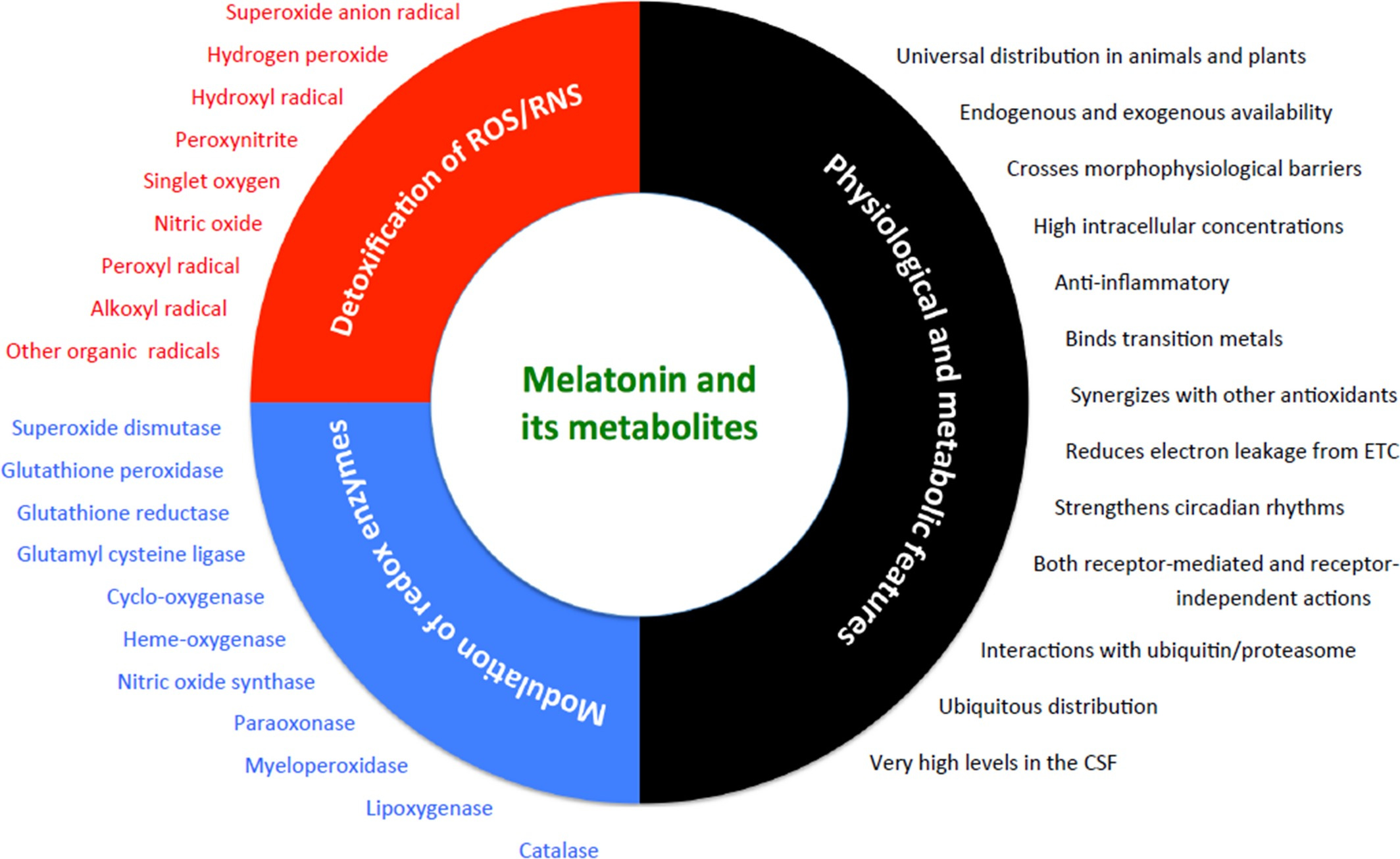
Lowes et al.176 compared the relative efficacies of the two mitochondria-targeted antioxidants, MitoQ and MitoE, with melatonin in reducing inflammation and oxidative damage.
A worst case scenario was used to create the molecular carnage. Adult male rats were given both bacteria lipopolysaccharide (LPS) and peptidoglycan (PepG) via a tail vein infusion to induce massive oxidative damage.
Shortly, thereafter, the animals received, via the same route, either MitoQ, MitoE or melatonin. Five hours later, plasma and tissue samples were collected. The authors described broadly equivalent protective actions of the three antioxidants relative to their improvement in maintaining mitochondrial respiration, reducing oxidative damage and depressing pro-inflammatory cytokine levels.
From the data in this figure, it seems apparent that the most effective antioxidant related to these parameters was melatonin given the lower mean values of damage molecules and their more uniform inhibition in the animals of this group.
The combination of LPS + PepG is a very aggressive challenge to the defensive makeup of mammals and in this study melatonin handled the attack as well as or better than the synthetic mitochondria-targeted antioxidants.
A major implication of these findings is that melatonin should be classified as an endogenous mitochondria-targeted antioxidant (Fig. 6). This would be consistent with the much higher melatonin levels in hepatic mitochondria than in the plasma as reported177, 178 and with the proposal that mitochondria might be the site of intracellular melatonin synthesis.179
Key takes from “Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics“38, (2016, emphasis mine) which builds on this work:
Melatonin has been speculated to be mainly synthesized by mitochondria. This speculation is supported by the recent discovery that aralkylamine N-acetyltransferase/serotonin N-acetyltransferase (AANAT/SNAT) is localized in mitochondria of oocytes and the isolated mitochondria generate melatonin.
It accumulates in mitochondria with high concentration against a concentration gradient. This is probably achieved by an active transportation via mitochondrial melatonin transporter(s).
Melatonin protects mitochondria by scavenging reactive oxygen species (ROS), inhibiting the mitochondrial permeability transition pore (MPTP), and activating uncoupling proteins (UCPs).
Thus, melatonin maintains the optimal mitochondrial membrane potential and preserves mitochondrial functions. In addition, mitochondrial biogenesis and dynamics is also regulated by melatonin.
Melatonin synthesis correlates to circadian patterns of mitochondrial activity in the pineal gland:
In most cases, melatonin reduces mitochondrial fission and elevates their fusion. Mitochondrial dynamics exhibit an oscillatory pattern which matches the melatonin circadian secretory rhythm in pinealeocytes and probably in other cells. Recently, melatonin has been found to promote mitophagy and improve homeostasis of mitochondria.
Serum levels are extremely low in comparison to the pineal gland and other tissues:
The physiological level of melatonin in serum of mammals is in the range of 10−9 M.
The physiological level of melatonin in the pineal recess of the third ventricle of sheep is at least 100-fold higher than that in the serum [87]. In unicellular organism, the physiological levels of melatonin reach 10−4 to 10−3 M [88]. As a result, it is difficult to distinguish the “physiological” levels of melatonin from pharmacological values depending on the tested fluid or tissue.
Melatonin promotes mitogenesis > promotes melatonin > inhibits ROS induced damage. Spike-induced ROS damages mitochondrial mtDNA and may reverse this cycle:
When male mice were exposed to constant light, not only was melatonin production depressed, but many pinealocyte mitochondria appeared swollen with a rarified matrix and reduced numbers of cristae [95]. These changes suggest that an additional function of mitochondria, besides ATP production, may be associated with melatonin synthesis.
It is our belief that, in addition to pinealocytes almost all organs, tissues and cells have the capacity to synthesize melatonin.
Different from the pinealocytes where melatonin is released into the blood and cerebrospinal fluid (CSF) as a signaling molecule to convey photoperiodic information [87,99], melatonin synthesized by other cells is presumably used locally for defense against oxidative stress and inflammation [100].
Interestingly, mitochondria seem not only to synthesize melatonin but also to metabolize it.
The first evidence of melatonin as a mitochondrial protector came from the report of Mansouri et al. [128]. The authors reported that melatonin attenuated the ethanol-induced hepatic mitochondrial DNA depletion in mice with the mechanism being related to melatonin’s antioxidant capacity.
Martin et al. [25] subsequently observed that melatonin prevented the inhibition of mitochondrial complexes I and IV induced by ruthenium red and significantly reduced mitochondrial oxidative stress caused by t-butyl hydroperoxide; however, comparable doses of vitamins C and E lacked these protective effects [111].
The differences among melatonin and vitamin C and vitamin E on the relative protection of mitochondria may be explained by the observations that melatonin accumulates in mitochondria perhaps via the active transport by PEPT1/2 (unpublished observations Ma et al.), but this is not the case with vitamin C and E.
…melatonin also exhibits significant beneficial effects on several neurodegenerative diseases related to mitochondrial dysfunctions per se. These include Huntington’s disease (HD), which is an autosomal dominant neurodegenerative disorder where the alterations in mitochondrial function play a key role in the pathogenic processes [145]. Melatonin administration significantly delayed disease onset and mortality in a transgenic mouse model of HD [146].
In a mouse model of MS, melatonin treatment prevented the pathological alterations by restoring mitochondrial respiratory enzyme activity and fusion and fission processes as well as by reducing intra-axonal mitochondria accumulation [148].
Moreover, a recent report showed that the treatment of a patient suffering with primary progressive MS exhibited significant clinical improvement after low-dose melatonin treatment [149]. The protective mechanisms of melatonin on mitochondria are multiple.
These include, but are not limited to, a reduction of mitochondrial oxidative stress [150,151], preservation of the mitochondrial membrane potential [152,153,154], upregulation of the antiapoptotic mitochondrial protein/downregulation of the proapoptotic mitochondrial protein, Bax [137,155,156], increased efficiency of ATP production [59,157], reduced release of cytochrome C into the cytosol and the inhibition of caspase 3 activity [158,159].
Many studies have addressed the importance of melatonin’s effects on the mitochondrial membrane potential (Δψ).
And from 2022, Cecon et al. used molecular modelling analysis and a mouse model to demonstrate how metformin can act as an antiviral against SARS-CoV-2 by inhibiting binding to ACE2. It would also ameliorate the effects of synthetic Spike, thus further protecting against mitochondrial damage and relates to my brain fog & long COVID Substack:39
Key takes from “Melatonin drugs inhibit SARS-CoV-2 entry into the brain and virus-induced damage of cerebral small vessels“40 (emphasis mine):
Brain infection is particularly pronounced in the K18-hACE2 mouse model of COVID-19. Prevention of brain infection in the acute phase of the disease might thus be of therapeutic relevance to prevent long-lasting symptoms of COVID-19.
We previously showed that melatonin or two prescribed structural analogs, agomelatine and ramelteon delay the onset of severe clinical symptoms and improve survival of SARS-CoV-2-infected K18-hACE2 mice.
Here, we show that treatment of K18-hACE2 mice with melatonin and two melatonin-derived marketed drugs, agomelatine and ramelteon, prevents SARS-CoV-2 entry in the brain, thereby reducing virus-induced damage of small cerebral vessels, immune cell infiltration and brain inflammation.
Molecular modeling analyses complemented by experimental studies in cells showed that SARS-CoV-2 entry in endothelial cells is prevented by melatonin binding to an allosteric-binding site on human angiotensin-converting enzyme 2 (ACE2), thus interfering with ACE2 function as an entry receptor for SARS-CoV-2.
Our findings open new perspectives for the repurposing of melatonergic drugs and its clinically used analogs in the prevention of brain infection by SARS-CoV-2 and COVID-19-related long-term neurological symptoms.
MLT10: 10 mg/kg melatonin.
MLT50: 50 mg/kg melatonin. High dose also used in mouse model as the half-life in plasma is only 20-30 minutes.
AgoMLT: Clinically used melatonin receptor ligand and structural analog agomelatin.
RML: Clinically used melatonin receptor ligand and structural analog ramelteon.
Collagen IV, Caveolin-1: endothelial marker proteins.
Empty string vessels are formed when either synthetic or viral Spike binds to ACE2 receptors in endothelial cells and causes their apoptosis. String vessels are arrowed:

Our study describes the first allosteric-binding site of ACE2 and melatonin is the first allosteric modulator of ACE2 which negatively regulates ACE2 binding to the SARS-CoV-2 spike protein. This opens additional opportunities for the development of ACE2-specific drugs that interfere with the spike interaction [81]. Importantly, binding of melatonin to this allosteric-binding site of ACE2 did not interfere with ACE2 enzyme activity, thus avoiding potential severe side effects as ACE2 activity is essential for the proper function of the renin–angiotensin system.
In conclusion, we disclosed ACE2 as a new binding target of melatonin, which leads to allosteric negative modulation of the spike/ACE2 interaction. This effect on the SARS-CoV-2 entry receptor function of ACE2, combined with the inhibition of NFKB expression and endogenous ACE2 expression in endothelial cells, is most likely the predominant mechanism for impaired SARS-CoV-2 brain invasion by melatonin and its derivatives.
SARS-CoV-2 and mitochondrial disfunction
ROS generation? Mitochondrial damage? New onset diabetes? MOTS-c depletion? Metabolic dysfunction? Cardiovascular damage? Neurological disorders and relapsing cases of MS and accelerated aging?
All this is eerily familiar, familiar as in being classical symptoms of long COVID (also known as post-acute sequalae of COVID-19, PASC).
 | Scoobypedia | Fandom")
Metformin provides a clue, it just remains to perform a literature review to link mitochondrial damage to both PASC and long term mRNA gene therapy injuries from the LNP/mRNA/Spike platform.
It’s important to note that MOTS-c depletion doesn’t mutually exclude other contributory factors, its just another piece in the puzzle. For example, BNT162b2 induced miR-21 expression suppresses a circadian clock protein called BMAL-1, and BMAL-1 shares many protective properties with MOTS-c, eg cardio-protection41, inhibition of diabetes42 and the aging process.43
The Humanin Touch
Similar to 12S, humanin (HN) is also a mitochondrial micropeptide with cytoprotective properties. The MT-RNR2 ribosomal RNA gene encoding humanin is also known as 16S. HN regulates multiple signalling pathways involved with oxidative stress.
Biological effects of HN include (From “Protective Mechanism of Humanin Against Oxidative Stress in Aging-Related Cardiovascular Diseases“, 2021):44
Increase of antioxidant enzyme and reduction of ROS (protects ovarian cells).
Restoration of cathepsin D function and promotion of autophagic degradation (inhibits lipid and cholesterol function; protection of aging cells).
Protection of osteoblasts from apoptosis (prevention of osteoporosis).
Reduction of ROS and intracellular calcium (protects the nervous system).
Reduction of ROS and NOX2 (cardiovascular protection).
Reduction of ROS and Improved glucose and lipid metabolism (inhibition of lipid accumulation; improved non-alcoholic fatty liver disease, NAFLD).
Reduction of ROS and inhibition of inflammation (inhibition of bone disorders).
Increase of antioxidant enzyme and reduction of ROS (protects the nervous system).
Other protective properties again include inhibition of T2DM through increased insulin sensitivity45 and inhibition of Alzheimer’s Disease through improved defence of neurons from beta-amyloid toxicity.46
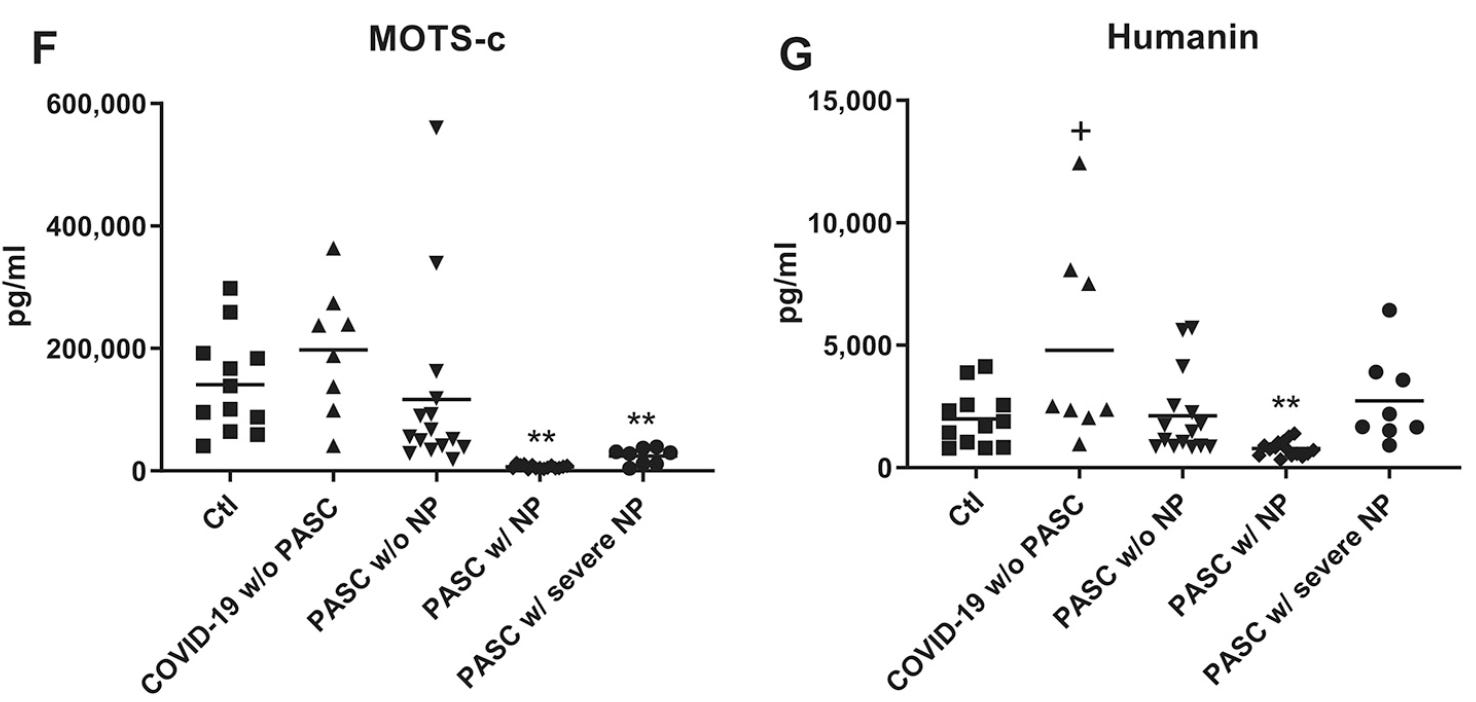
From 2023“Evaluation of Serum Humanin and MOTS-c Peptide Levels in Patients with COVID-19 and Healthy Subjects”47 by Saracaloglu et al enrolled 38 adult COVID-19 patients, with 32 gender-matched healthy adult control volunteers.
They found that both MOTS-c and humanin were involved in the pathogenesis of COVID-19. Serum levels of humanin were significantly depressed but MOTS-c was elevated, possibly as a compensatory mechanism. Antiviral treatments had no effect on serum MOTS-c and humanin levels:


Of relevance here, vaccinated patients were excluded from the study:
“Patients receiving a COVID-19 vaccine, having an unconfirmed diagnosis with the negative viral RNA in RT-PCR results, diagnosed hepatic, cerebrovascular, and renal diseases, neoplasia, pregnant or breastfeeding women, the requirement for extracorporeal membrane oxygenation, intensive care unit, or mechanical ventilation were excluded from the study.”
A study from the previous year also excluded the vaccinated, but considered correlations with long COVID and neuropsychiatric symptoms too.
“SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19”48 by Peluso et al enrolled 12 controls and 46 patients with combinations of COVID-19, PASC and neuropsychiatric manifestations (NP):
Control: 12
COVID-19 w/o PASC: 8
PASC w/o NP: 15
PASC w/ NP: 15
PASC w/ severe NP: 8
The most disabling PASC symptoms are respiratory and include “dyspnea, chest pain, cough and wheezing”.
NP symptoms include “attention deficit, memory loss, depression, confusion, anxiety, obsessive–compulsive disorders and special sensory impairments.”
NDEV: neuron-derived extracellular vesicles, in this case transporting SARS-CoV-2 proteins and mitochondrial proteins (MPs).
ADEV: astrocyte-derived EVs, a subtype of glial cells that make up the majority of cells in the human CNS.49
What they found was that both humanin and MOTS-c all were significantly lower in patients with both both PASC and NP but not with PASC without NP when compared to controls.
NDEV levels were lower in PASC with and without NP, and ADEV levels of calcium channel MPs mitochondrial calcium uniporter (MCU) and sodium/calcium exchanger (NCLX) were also increased in PASC with and without NP.
Key takes (emphasis mine):
The mean levels of both S1 (RBD) and N proteins were significantly higher in NDEVs than ADEVs of the PASC w/ NP subgroup, which implied higher intracellular and possibly intramitochondrial levels in neurons than astrocytes.
MCU and NCLX are the principal channels for moving Ca++ in and out of the mitochondrial matrix, respectively, and the Na++/Ca++ exchanger LETM1 is one of many alternative channels to MCU.
The importance of normal Ca++ efflux from mitochondria for neuronal function was demonstrated by the metabolic abnormalities, including increased levels of ROS, that resulted when expression of NCLX was genetically reduced. A similar reduction in the expression of NCLX in neurons increased the rate of CNS accumulation of amyloid and altered behavior in a mouse model of AD.
Exclusion of vaccinated patients is unfortunate, but as we know from prior studies that both LNPs and cleaved vaccinal Spike S1 can readily cross the blood brain barrier the study itself gives us a high confidence in our hypothesis:
As for the mitochondrial proteins, it is not possible yet to assume that NDEV levels of SARS-CoV-2 proteins accurately reflect those in the central nervous system. Nonetheless, the mean levels of both S1 and N were significantly higher in ADEVs and NDEVs of all affected subgroups than background levels in controls. Further, mean ADEV and NDEV levels of N distinguished PASC w/NP from PASC w/o NP and from COVID-19 w/o PASC.
Both N protein (viral only) and Spike S1 extracellular vesicle levels correlated strongly with PASC and NP outcomes, as well as MOTS-C and humanin serum mitochondrial protein. As PASC symptoms can present for months or years after infection and/or transfection then this implies persistent expression and caution warranted against any reinfection, even with so called “mild” Omicron:

In summary, Spike S1/N protein expression leads to a cascade of events, involving but not limited to:
Elevation of pro-inflammatory cytokines, including due to widespread lipopolysaccharide (LPS) endotoxin contamination of synthetic mRNA batches.50
A disrupted renin-angiotensin-aldosterone system (RAAS), including depletion of angiotensin-converting enzyme (ACE-2, antioxidant)51 and overexpression of angiotensin II (ANG II, oxidant).52 ACE-2 binding in turn can induce more IL-6.53
Damage to mtDNA.56
MOTS-c/humanin and melatonin depletion due to either cellular apoptosis or autophagy. Diabetes may result if this response is dysfunctional.57.
Autoimmune disorders and more oxidative stress related disorders - both neurological and cardiovascular, including increased insulin resistance.58
As mitochondria can both generate ROS and be destroyed by it (due to apoptosis or mitophagy, as above) the cycle may repeat either locally or systemically until enough have been destroyed and mitochondrial homeostasis is restored.59

From: “Model for IL-6-mediated oxidative stress reduction in β-cells. IL-6 signaling activates autophagy, leading to p62-mediated degradation of the antioxidant repressor KEAP1. Decreased KEAP1 allows the accumulation of NRF2, which translocates to the mitochondria, and mitophagy is stimulated to reduce ROS. Together, these events protect β-cells from oxidative stress and promote β-cell survival. MMP, mitochondrial membrane potential; p-p62, phosphorylated p62.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6054440/
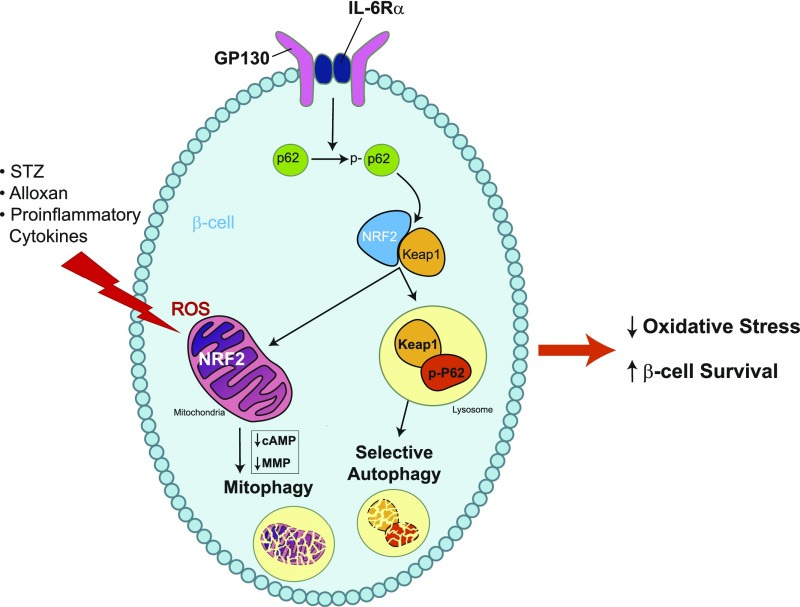
It should also be noted that the LNP platform itself elevates proinflammatory cytokines including IL-660 which will further contribute to mitochondrial damage.61 62
SARS-CoV-2 and mitochondrial gene down-regulation
Guarnieri et al. (2023) published “Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts”.63 It has a co-authorship totalling 51 - almost rivalling the supporting cast of Ben Hur:


We found that at peak infection time, there are distinct changes in different regions of the brain, including is a large decrease in mitochondrial genes in the cerebellum, the part of the brain that controls our muscles, balance, cognition, and emotion. The lung is the primary site of infection, but molecular signals are being transmitted affecting the entire body, with the heart, kidney, and liver being more affected than others, even long after the virus is gone."
Jonathan C. Schisler, PhD, assistant professor of pharmacology and member of the UNC McAllister Heart Institute (2023)
The full study runs to 19 pages. A full review would need its own Substack, but really we only need to focus on the viral or viral protein interactions.
Unsurprisingly there are a couple of conflicts of interest declarations, but the biggest red flag is part funding by the Bill & Melinda Gates Foundation.
Key takes:
They took around 700 nasopharyngeal samples, both infected and uninfected controls from patients in New York City from 2018-20. The study was published long after the vax had been rolled out.
Samples were divided into low, medium and high SARS-CoV-2 viral loads and transcription analysis performed.
They found that the virus inhibited all 5 mitochondrial oxidative phosphorylation (OXPHOS, ie ATP generation) complexes.
This was associated with changes to mitochondrial form, including matrix condensation and swollen cristae, a reduction in inner membrane protein import systems and increased levels of mitochondrial ROS.
The virus upregulated miRNA-2392 - activating hypoxia-inducible factor-1a (HIF-1a, a major regulator of oxygen homeostasis within cells) and inducing glycolysis.
Autopsy tissue samples found that even though the lungs were free of viral mRNA and mitochondrial gene expression function had recovered this wasn’t the case with the heart tissues, and less so with the kidneys and liver.
A study using infected hamsters found that at peak viral load the lungs were minimally affected but the cerebellum was down-regulated and the striatum up-regulated.

Striatum in red, amygdala in pink, thalamus in blue. Licensed under the Creative Commons Attribution-Share Alike 4.0 International license. https://upload.wikimedia.org/wikipedia/commons/f/f2/Striatum.svg The authors suggest that after the viral titres have peaked mitochondrial gene expression is suppressed, glycolysis induced and antiviral immune responses are triggered. Although this clears the virus, mitochondrial dysfunction persists in the lymph nodes, heart, kidney and liver. This all contributes to severe COVID-19 pathologies.
Viral polypeptides bind to host polypeptides. Around 16% of these are mitochondrial:
M protein binds to coenzyme Q (CoQ) synthesis protein COQ8B, asparaginyl-tRNA (NARS2) synthases and mitochondrial leucyl-tRNA (TARS2).
N protein interacts with several complex I subunits and import peptidases PMPCA plus PMPCB.
Complex V subunits bind to viral nonstructural protein NSP6.
Viral open reading frame ORF10 binds to TIMM8.
ORF9b binds to TOMM70.

This is where antioxidants including NAC, co-enzyme Q10 and its synthetic analogue MitoQ are of value:
“…treatment with the ROS scavengers N-acetyl cysteine (NAC) and MitoQ reduced HIF-1α, glycolytic proteins, pro-inflammatory cytokine mRNAs, and viral load.”
Bearing in mind one of the key sponsors it is curious that effects of a protein common to both virus and synthetic mRNA expression is not referred to directly, not even once. This being Spike protein of course. It is only cited indirectly, yet we know from binding to antioxidant ACE2 and RAAS disruption it is a major source of mitochondrial ROS and mtDNA damage.

“SARS-CoV-2–infected mice expressing the human angiotensin-converting enzyme 2 receptor (K18-hACE2 mice) have reduced nDNA- and mtDNA-encoded mitochondrial OXPHOS proteins in the heart, lung, kidney, and spleen (18).”64
They move swiftly on in the discussion, don’t look here, look there!

Not so fast! I will help the authors here a little:

Almost all the mitochondrial OXPHOS genes were deactivated. If the virus (or synthetic Spike for that matter) reaches your heart it can put it under metabolic stress:
“…table S1) confirmed that virtually all OXPHOS mRNAs of the heart were shut down (Fig. 2B). This was not simply the product of terminal destruction of heart cells because mtDNA transcripts and the COX assembly gene mRNAs, COX16, COX19, and COX20, were up-regulated and PET100 and SCO2 were normally expressed (Fig. 2B). The OXPHOS mRNA profiles in the COVID-19 autopsy samples of the kidney and liver (Fig. 2, B and C) were similar.”

They don’t say it but Spike has been found to persist in the heart and lymph nodes for months after gene transfection, and more evidence of reverse transcription AND expression is emerging.65,66
“These results suggest that SARS-CoV-2 mRNA vaccines routinely persist up to 30 days from vaccination and can be detected in the heart.”
From: “Duration of SARS-CoV-2 mRNA vaccine persistence and factors associated with cardiac involvement in recently vaccinated patients“ (2023)
…Blood samples were collected from a cohort of 81 patients experiencing long-COVID symptoms.
…PCR (Polymerase Chain Reaction) was performed using specific primers designed to target the spike protein sequence derived from the BNT162b2 vaccine (BioNTech/Pfizer mRNA Vaccine) (1). The forward primer (CGAGGTGGCCAAGAATCTGA) and reverse primer (TCTGGAACTAGCAGAGGTGG) were used.
…The PCR products obtained from the Nested PCR were subjected to Sanger sequencing to determine the nucleotide sequence of the amplified fragment.
…However, it is essential to acknowledge the limitations of this study, including the short length of aligned sequences and primer-related constraints, which necessitate further investigation to confirm vaccine integration and exclude potential cross-reactivity or contamination (1).
https://www.europeanreview.org/wp/wp-content/uploads/supplementary-data.pdf
…This study, in agreement with other published investigations, demonstrates that both natural and vaccine spike protein may still be present in long-COVID patients, thus supporting the existence of a possible mechanism that causes the persistence of spike protein in the human body for much longer than predicted by early studies. According to these results, all patients with long-COVID syndrome should be analyzed for the presence of vaccinal and viral spike protein.
From: “Presence of viral spike protein and vaccinal spike protein in the blood serum of patients with long-COVID syndrome“ (2023)

“Down-regulation of mitochondrial pathways in response to virus in human autopsy samples was most extensive in hearts followed in decreasing order by the kidneys, livers, and lymph nodes (fig. S5). This inhibition of mitochondrial gene expression in visceral autopsy tissues parallels that seen in the nasopharyngeal samples with high viral titer but in the absence of a detectable virus.”
Although it was a mammoth, well funded production that would rival some of the Hollywood greats we must shout “Cut!” here.

Indeed, because they assayed every viral protein apart from THE most relevant and cytotoxic one the study feels almost half baked and compromised, and we will come to that next.
Spike protein impairment of cardiac mitochondria
I recommend reading Walter’s review work on this too as we often focus on different aspects.67
Huynh et al. published “Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19”68 in 2023. They used a Seahorse XFe24 extracellular flux analyser to study mitochondrial bioenergetics changes of cardiomyocytes in real time in response to 1nM Spike S1 exposure for 24, 48 and 72h. S1 exclusion was used as a control, and no seahorses were harmed.
ROS production, mitochondrial Ca2+ levels and expression of proteins linked to OXPHOS and signalling cascades were evaluated by using Western blotting, XRhod-1, and MitoSOX Red staining.



They found that OXPHOS increased for the first 24 hours, then decreased after 72 h. This was associated with increased ROS, mitochondrial and intracellular calcium, increased mitochondrial fragmentation/fission and a negative mitochondrial membrane potential. This is not good at all.
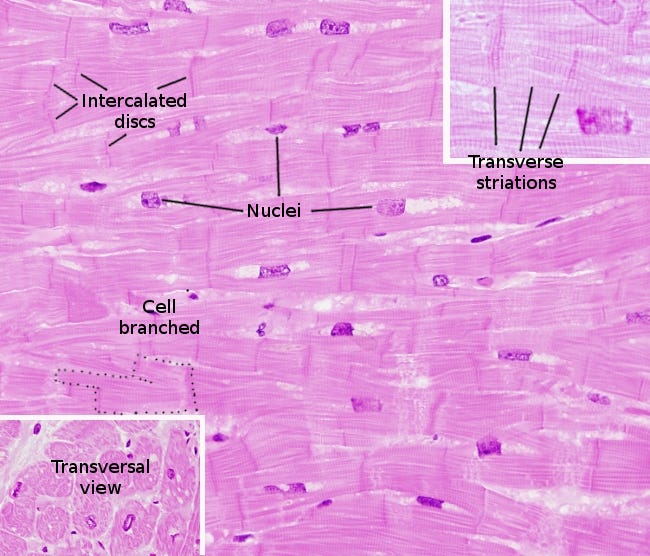
Cardiomyocytes, or cardiac muscle cells undergo involuntary contraction, are striated, branched and contain a large number of mitochondria to power them.69
Mitochondrial membrane potential (Δψm, pronounced “delta-sy-em”, also known as MtMP) is used to indicate mitochondrial activity, as the charge differential caused by anion accumulation is linked to ATP production, the electron transport chain (ETC) and OXPHOS.
…Mitochondria constantly divide, fuse, and alter their size and shape, forming a dynamic network to maintain their integrity and quantity. Normal mammalian cells maintain a balance between fusion and fission.

…A positive mitochondrial membrane potential (Δψm) of 120‐200 mV is fundamental for the normal performance and survival of cells, especially those that have a high‐energy requirement. Thus, loss of Δψm is an indicator of reduced cell health. The collapse of Δψm due to the opening of a high‐conductance pore in the inner mitochondrial membrane is part of the molecular mechanism in apoptosis. The mitochondrial uncoupler carbonyl cyanide‐m‐chlorophenylhydrazone (CCCP) depolarizes the inner mitochondrial membrane, reducing Δψm and ATP production, thus increasing the level of AMP and the phosphorylated (active) AMP‐activated protein kinase (pAMPK). This sequence of events elevates the level of reactive oxygen species (ROS), leading to oxidative damage.
…Impairment of mitochondrial dynamics can result in reduced oxidative phosphorylation and cell death. Mitochondria produce ATP for cellular energy requirements, and ROS are toxic by‐products generated during OXPHOS in mitochondria. Mitochondria generate about 90% of the ROS present in cells. Excess ROS may cause oxidative damage to both nuclear DNA (nDNA) and mtDNA, which may contribute to many age‐related disease states.29 Compared to nDNA, mtDNA is prone to be damaged by oxidative stress because it is not protected by associated histones and other chromatin proteins and is near the ROS‐generating respiratory chain.
…Besides ROS, replication errors and failure of the repair mechanisms may be the more important reason for accumulation of mtDNA mutations.30 mtDNA replication is independent of cellular division, so the replication rate is higher than that of nDNA.31
…Mitochondrial dysfunction occurs in most neurodegenerative diseases, including Parkinson's diseases (PD), Alzheimer's (AD), Huntington's disease (HD), Friedreich's ataxia (FRDA), and amyotrophic lateral sclerosis (ALS).
From: “Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure“ (2019)
There is a fantastic gif available via the “Small Things Considered” website.70
Rather than the unchanging, static “beans” we see in all the textbooks they are a lot more amorphous than that and behave more like bacteria, a clue to their probable origins.
They lack flagella but adaptor proteins may attach them to motor proteins and drag them along actin filaments or microtubules in the cytoplasm, occasionally fusing or splitting as they go.
I’ve used an animation of a motor protein before, and its worth including again. These guys are the unsung heroes of our cells, toiling away 24/7 for nothing more than ‘payment’ in ATP:

It took 10 minutes to record this excellent high magnification looping time lapse of mitochondria being dragged along:
Impaired mitochondrial Ca2+ uptake can affect the cytosolic concentration of Ca2+ [Ca2+]c, due to pathological signalling and downregulation of mitochondrial metabolism. This can, in itself, lead to the death of cardiomyocytes:
Under pathological conditions of cellular [Ca2+]c overload, particularly in association with oxidative stress, mitochondrial Ca2+ uptake may trigger pathological states that lead to cell death. In the model of glutamate excitotoxicity, microdomains of [Ca2+]c are apparently central, as the pathway to cell death seems to require the local activation of neuronal nitric oxide synthase (nNOS), itself held by scaffolding proteins in close association with the NMDA receptor. Mitochondrial Ca2+ uptake in combination with NO production triggers the collapse of mitochondrial membrane potential, culminating in delayed cell death.

From: “Mitochondria and calcium: from cell signalling to cell death“ (2020)
Key takes from the study (emphasis mine):
The 24 h S1 treatment increased ATP production and mitochondrial respiration by increasing the expression of fatty-acid-transporting regulators and inducing more negative mitochondrial membrane potential (Δψm).
The 72 h S1 treatment decreased mitochondrial respiration rates and Δψm, but increased levels of reactive oxygen species (ROS), mCa2+, and intracellular Ca2+.
Electron microscopy revealed increased mitochondrial fragmentation/fission in AC16 cells treated for 72 h.

Mito-Tempo is mitochondria-targeting antioxidant. It can enhance the effects of NAC and be administered to patients to reduce the risk of paracetamol overdose induced liver damage (acetaminophen induced hepatotoxicity, APAP). NAC is currently the only available treatment for APAP.71
Put another way it takes some pretty strong overdose treating emergency antioxidants to even start to protect your cells from Spike S1 induced toxicity.
I don’t remember hearing that at any of the coercive press conferences…

The effects of S1 on ATP production were completely blocked by neutralizing ACE2 but not CD147 antibodies, and were partly attenuated by Mitotempo (1 µM).

Conclusion: S1 might impair mitochondrial function in human cardiomyocytes by altering Δψm, mCa2+ overload, ROS accumulation, and mitochondrial dynamics via ACE2.
Cardiac injury is the second most prevalent manifestation of COVID-19 in hospitalized patients (20–30%) [1] and is associated with disease severity and associated mortality [1,2,3].
Cardiac injury includes acute complications and various post-acute sequelae, such as myocarditis, ischemic heart disease due to hypercoagulation [1], microvascular dysfunction, cardiac vasculitis of small vessels [4], arrhythmias, and heart failure.
The reduction of the ATP synthesis capacity of cardiac mitochondria represents the key mechanism underlying cardiac contractile dysfunction [14,15].
They not only serve as the “power house” of cells, but also regulate cardiac ion homeostasis, cellular growth and apoptosis, protein quality, inflammation, and redox balance [16]. Therefore, in addition to the reduction of ATP synthesis, mitochondrial dysfunction impairs various functions, ultimately leading to cardiac diseases.
Ca2+ is involved in various cellular regulatory mechanisms. mCa2+ exerts biphasic effects on energetics, cardiac function, and oxidative balance. Low mCa2+ levels may result in the tricarboxylic acid cycle failing to generate enough reduced fuel substrates to match the rate of oxidation in the mitochondrial electron transport chain (ETC), leading to a redox imbalance.
…an mCa2+ overload leads to the excessive production of reducing equivalents and redundant delivery of electrons to ETC, which promotes ROS production [37,38]. Furthermore, excessive mCa2+ may induce the formation of mitochondrial permeability transition pores, thus, disrupting Δψm and causing mitochondrial dysfunction [39,40].
The activity of ATP synthase (complex) is maintained by the proton (H+)-driving force, which is a combination of Δψm and the H+ gradient. Thus, any change in Δψm impairs mitochondrial function.
This also leads to ROS production:
In the range of optimized Δψm, mROS production is maintained at physiological levels; extreme high or low Δψm may induce the overproduction of mROS. Through MitoSOX Red staining, we found that AC16 cells treated with S1 for 72 h exhibited higher levels of ROS than control cells; this might have been because S1 substantially disrupted Δψm and increased mROS production. In turn, the accumulation of ROS exacerbated mitochondrial dysfunction.
The Mitotempo pretreatment could partly attenuate the impact of S1 on mitochondrial respiration. This result further confirmed the role of ROS in mitochondrial dysfunction, at least in part. Accordingly, mitochondrial antioxidants are the potential agents to reduce the detrimental effects of S1 by oxidative stress.
Using MitoTracker confocal fluorescence microscopy, we observed that mitochondrial fragmentation and network disruption increased in S1-treated (72 h) AC16 cells compared with control cells. Furthermore, TEM revealed that the S1 treatment for 72 h induced mitochondrial fission in AC16 cells, as evidenced by the decrease in size and increase in the number of mitochondria compared with the findings for control cells.
The binding of S1 to the ACE2 receptor initiates the cleavage of ACE2 at the ectodomain and endodomain through the host’s metallopeptidase domain 17 and transmembrane protease serine 2. This process leads to the shedding and downregulation of ACE2 [44,45].
Therefore, SARS-CoV-2 infection decreases ACE2 on the cell membrane and their activity, and increases Ang II-mediated pathophysiology.
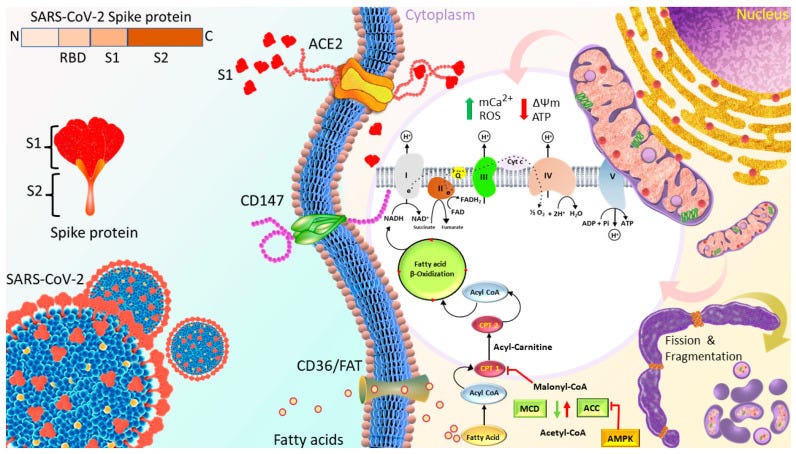
The authors finish with a somewhat sobering conclusion*. This is all the more startling given that these findings were published 2+ years after the mass administration of efficient S1 expressing gene agents:
To the best of our knowledge, this is the first study to evaluate the effects of S1 on mitochondrial function in human cardiomyocytes. Although S1 improved mitochondrial function in AC16 cells in the short term, prolonged S1 treatment led to mitochondrial dysfunction due to the disruption of Δψm, mitochondrial Ca2+ overload, accumulation of ROS, and alteration of mitochondrial dynamics. These effects of subunit S1 of the S protein translate into irreversible cardiac remodeling.
*Cardiac remodeling is defined as a group of molecular, cellular and interstitial changes that manifest clinically as changes in size, mass, geometry and function of the heart after injury. The process results in poor prognosis because of its association with ventricular dysfunction and malignant arrhythmias.
Cardiac dysfunction is the main consequence of cardiac remodeling, which consists of a pathophysiological substrate for the onset and progression of ventricular dysfunction. This interaction starts with genetic changes in response to a cardiac injury, with reexpression of fetal genes. Consequently, cellular and molecular changes occur, resulting in progressive loss of ventricular function, asymptomatic at first, that evolves to signs and symptoms of heart failure4-6,9 (Figure 2).

From: “Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment“ (2016)
A lot of victims may be at one of these stages, perhaps asymptomatically at present and have no idea until, say ten years later they are carrying a heavy bag of shopping in and suffer a fatal cardiac arrest without warning in middle age.
Krauson et al. (2023)72 used RT-qPCR-based assays to detect BNT162b2 and mRNA-1273 in the lymph nodes, liver, spleen and myocardium from deceased patients. They detected vaccine in axillary lymph nodes in the majority of patients dying within but not more than 30 days from vaccination.
It was also found in the myocardium of a subset of patients vaccinated within 30 days of death, and myocardial healing associated macrophages were in greater number in the vaccinated than unvaccinated.
Traces of synthetic mRNA were not detected in the mediastinal lymph nodes, spleen, or liver:

They concluded from their results “that SARS-CoV-2 mRNA vaccines routinely persist up to 30 days from vaccination and can be detected in the heart.”
The next question we have is “once either COVID-19 infection or synthetic mRNA gene agents have damaged mitochondrial function how long do they take to recover? A week? 30 days maybe?”
If we use loss of mitochondrial membrane potential (ΔΨ m) as a proxy for mitochondrial damage and loss of function, then a study from 2022 by Diaz-Resendiz et al. goes a long way to answering that. And their findings are alarming as ΔΨ m was significantly decreased in both males and females for 40 days +-13, and at least 11 months post infection in females vs healthy controls. This may be a factor in long-COVID pathologies, probably correlates with vaccinal mRNA and Spike expression and anecdotally may persist for at least 2-3 years.
Impaired leucocyte (white blood cells) function due to mitochondrial dysfunction is associated with oxidative stress and systemic inflammation.73 Spike S1 has motifs homologous to the glycosylated envelope protein gp120 of HIV, which is also a contributory factor to the early CD4+ T cell depletion of AIDS patients. I go into more detail in my “brain fog” Substack.74

Live virus is not a necessity, and Spike has a strong binding affinity for CXCR-4 and CCR5 (=CD195) too.75,76
The first column in green depicts experimentally predicted binding of Spike to CCR5 co-receptors on CD4+ T-cells:

Let us consider this mechanism using the Env glycoprotein as an example, which is cleaved into 2 proteins during maturation: gp41 and gp120. The key role in the initiation of apoptosis is played by gp120, which is associated with interactions with co-receptors of CD4+ T-lymphocytes, CXCR4 and CCR5. This interaction ensures the penetration of HIV into the target cell. At the same time, it has been shown that gp120 is able to induce mitochondria-mediated apoptosis by depolarizing the mitochondrial membrane, which primarily leads to a disruption in the process of oxidative phosphorylation and, as a result, also leads to a decrease in ATP production and the energy depletion of T-lymphocytes, finally leading to cell death.
…Due to its ability to bind to cellular receptors, localize to the cytoplasmic membrane of an infected cell and transfer to an adjacent uninfected cell, Env can activate mitochondrial dysfunction and lymphocyte apoptosis without direct infection with virions, which is one of the explanations for the rapid decline in CD4 + T-lymphocytes at an early stage of infections [36].
From: “Impaired Mitochondrial Function in T-Lymphocytes as a Result of Exposure to HIV and ART“ (2023)
Key takes from “Loss of mitochondrial membrane potential (ΔΨ m) in leucocytes as post‐COVID‐19 sequelae”77.
Patients were classified into 4 groups:
(1) group of HC subjects: negative for SARS‐CoV‐2 and any symptoms or signs related to COVID‐19 were declared (n = 35).
(2) group of patients with COVID‐19 (C‐19): subjects with SARS‐CoV‐2 active infection (qRT‐PCR positive) (n = 36).
(3) group of subjects recently recovered from COVID‐19 (R1): individuals who were diagnosed as SARS‐CoV‐2 positive in our laboratory facilities, come back after several weeks of recovery (40 ± 13 dai), and resulted negative to SARS‐CoV‐2 molecular tests (n = 34).
(4) group of recovered subjects at 11 months post‐COVID‐19 (R2): individuals who were diagnosed as SARS‐CoV‐2 positive in our laboratory facilities, come back after 335 ± 20 dai (n = 18).
Patients who smoked or those with comorbidities (asthma, diabetes, hypertension, and neurodegenerative diseases) were excluded.
The results of this research, show that the ΔΨ m decreases significantly in subjects C‐19, R1, and R2, with respect to HC (Figure 1), suggesting that ΔΨ m in HPBMC is altered by acute SARS‐CoV‐2 infection and long‐COVID. These results align with previous reports by other authors, who indicate that SARS‐CoV‐2 causes mitochondrial dysfunction, since SARS‐CoV‐2 infection alters mitochondrial functionality, influencing its intracellular survival and/or evading host immunity. 4 , 17 , 26 , 27 , 28

From: “FIGURE 1. Mitochondrial membrane potential (MFI of DIOC6(3)) of HPBMC from healthy controls (HC) subjects, patients with COVID‐19 (C‐19), subjects recently recovered from COVID‐19 (R1) at 40 ± 13 days after infection (dai) and recovered subjects at 11 months post‐infection (R2, at 335 ± 20 dai). (A) Representative plots of ΔΨ m in HPBMC analyzed with software FlowJo. Each histogram shows DIOC6(3) MFI, which has affinity for stable ΔΨ m in cells. (B) MFI was represented as a bar graph with the means ± sem. Nonparametric Kruskal–Wallis and Dunn's multiple comparison tests were performed”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9088601/figure/jlb11111-fig-0001/

Recovery was partial for the R2 group and 15 of the 18 still had ongoing symptoms associated with long COVID, although cardiac scans were not performed or included:
…These results suggest that the loss of ΔΨ m in subjects recovered 335 ± 20 days after SARS‐CoV‐2 infection can be associated with the so‐called long‐COVID, since of the 18 individuals who participated, 85% reported symptoms such as weariness, difficulty sleeping, fatigue, dyspnea, memory problems, anxiety, arthralgia, headache, dry cough, chest pain, myalgia, and anosmia (Figure 2).

From: “FIGURE 2. Symptomatology classification in male (left side) and female (right side) patients with COVID‐19 (C‐19), subjects recently recovered from COVID‐19 (R1) at 40 ± 13 days after infection (dai) and recovered subjects at 11 months post‐infection (R2, at 335 ± 20 dai). The intensity of blue color in each box indicates the frequency of individuals who presented each symptom.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9088601/figure/jlb11111-fig-0002/ In this regard, some authors have reported that patients who manage to survive the effects of acute SARS‐CoV‐2 infection may develop long‐term sequelae, such as lung injury (pulmonary fibrosis), neuronal injury (acute and chronic neuropathology), and neurodegenerative diseases (Alzheimer's, Parkinson's and multiple sclerosis), 14 , 15 , 16 , 29 , 30 which could be attributed to mitochondrial dysfunction associated to SARS‐CoV‐2 infection, since it has been suggested that mitochondrial dysfunction in conjunction with an abnormal innate immunity response plays a pivotal role in the onset and development of several chronic diseases. 25 , 26 , 27 , 30 , 31 , 32 , 33
In general, the results indicate that female HC have higher ΔΨ m than male HC (Figure 3). In this sense, some authors report that between both sexes there are differences in mitochondrial function, in this way, compared with men, women present higher levels of enzymes (citrate synthase activity) ATP, and antioxidant compounds, 34 , 35 due to a higher mitochondrial activity, facts related to a longer life expectancy, 36 and a lower occurrence of diseases and aging. 37

From: “Mitochondrial membrane potential (MFI of DIOC6(3)) of HPBMC from healthy controls (HC), subjects, patients with COVID‐19 (C‐19), subjects recently recovered from COVID‐19 (R1) at 40 ± 13 days after infection (dai) and recovered subjects at 11 months postinfection (R2, at 335 ± 20 dai). MFI was represented as a bar graph with the means ± sem. The table shows the statistically significant differences between male and female. Nonparametric Kruskal–Wallis and Dunn's multiple comparison tests were performed”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9088601/figure/jlb11111-fig-0003/


The authors also note that despite females suffering a greater long term decline in ΔΨm than males, post-SARS-CoV-2 mitochondrial associated mortality was actually greater in males than females.
Spike induced autoantibodies to mitochondria must also be considered. Antimitochondrial M2 antibodies are associated with primary biliary cirrhosis (or primary biliary cholangitis, PBC), which can eventually lead to liver failure.78

In case you wondering, yes, there are case reports.
The inexcusable outcome from this study was not to strongly advise against any further boosters, but just to alternate future booster types.
Have these people no shame? Are they conflicted or just cretins?
The patient explained that he had developed jaundice, fatigue, and tiredness which had lasted a week and settled, and hence he did not seek any further medical attention. He further disclosed that he had similar symptoms nine months prior, that only lasted three days, and similarly did not seek medical attention. He further recalled that each episode had occurred following the COVID-19 vaccination. Initially, he was vaccinated with AstraZeneca/Oxford COVID-19 (ChAdOx1 S {recombinant} nine months before (March 2021). Following this, he was given the same vaccine again three months later (June 2021), and finally, he took a booster (Pfizer-BioNTech) COVID-19 vaccine (January 2022). He explained the symptoms of jaundice occurred for a few days each time following the COVID-19 vaccination. Liver screen test results were negative. The estimated non-invasive, updated Roussel Uclaf Causality Assessment Method scale (RUCAM) was 1 (Table (Table3)3) [17].

…The patient agreed to undergo a liver biopsy which demonstrated features compatible with a drug-induced cholangiopathy.
Subsequent liver function tests completely normalized with conservative treatment. The patient was advised to alternate COVID-19 vaccines for future boosters. Repeat LFTs at six months were normal.
From: “COVID-19 Vaccination-Induced Cholangiopathy and Autoimmune Hepatitis: A Series of Two Cases“ (2022)

Disclaimer
This site is strictly an information website about pathology, potential therapeutic agents and a review of the current state of research. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
References
Pizzorno J. Mitochondria—Fundamental to Life and Health. Integr Med (Encinitas). 2014;13(2):8-15. Accessed December 3, 2023. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4684129/
Zhang L, Zhang W, Li Z, et al. Mitochondria dysfunction in CD8+ T cells as an important contributing factor for cancer development and a potential target for cancer treatment: a review. J Exp Clin Cancer Res. 2022;41:227. doi:10.1186/s13046-022-02439-6
Lin YH, Lim SN, Chen CY, Chi HC, Yeh CT, Lin WR. Functional Role of Mitochondrial DNA in Cancer Progression. Int J Mol Sci. 2022;23(3):1659. doi:10.3390/ijms23031659
Saha T, Dash C, Jayabalan R, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. 2022;17(1):98-106. doi:10.1038/s41565-021-01000-4
Baldwin JG, Gattinoni L. Cancer cells hijack T-cell mitochondria. Nat Nanotechnol. 2022;17(1):3-4. doi:10.1038/s41565-021-01006-y
Dilna A, Deepak KV, Damodaran N, et al. Amyloid-β induced membrane damage instigates tunneling nanotube-like conduits by p21-activated kinase dependent actin remodulation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2021;1867(12):166246. doi:10.1016/j.bbadis.2021.166246
Pepe A, Pietropaoli S, Vos M, Barba-Spaeth G, Zurzolo C. Tunneling nanotubes provide a route for SARS-CoV-2 spreading. Science Advances. 2022;8(29):eabo0171. doi:10.1126/sciadv.abo0171
Valdebenito S, Ono A, Rong L, Eugenin EA. The role of tunneling nanotubes during early stages of HIV infection and reactivation: implications in HIV cure. NeuroImmune Pharmacology and Therapeutics. 2023;2(2):169-186. doi:10.1515/nipt-2022-0015
Dodagatta-Marri E, Meyer DS, Reeves MQ, et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer. 2019;7:62. doi:10.1186/s40425-018-0493-9
Seibel P, Trappe J, Villani G, Klopstock T, Papa S, Reichmann H. Transfection of mitochondria: strategy towards a gene therapy of mitochondrial DNA diseases. Nucleic Acids Res. 1995;23(1):10-17.
Koczor CA, Snyder JW, Shokolenko IN, Dobson AW, Wilson GL, Ledoux SP. Targeting repair proteins to the mitochondria of mammalian cells through stable transfection, transient transfection, viral transduction, and TAT-mediated protein transduction. Methods Mol Biol. 2009;554:233-249. doi:10.1007/978-1-59745-521-3_15

Martinez-Lopez JE, Coleman O, Meleady P, Clynes M. Transfection of miR-31* boosts oxidative phosphorylation metabolism in the mitochondria and enhances recombinant protein production in Chinese hamster ovary cells. J Biotechnol. 2021;333:86-96. doi:10.1016/j.jbiotec.2021.04.012
Quantum microRNA Assessment of COVID-19 RNA Vaccine: Hidden Potency of BNT162b2 SASR-CoV-2 Spike RNA as MicroRNA Vaccine| Crimsonpublishers.com. Accessed December 11, 2023. https://crimsonpublishers.com/aics/fulltext/AICS.000552.php
MIR21 (microRNA 21). Accessed December 11, 2023. https://atlasgeneticsoncology.org/gene/44019/mir21-(microrna-21)
Wu H, Wang J, Ma H, Xiao Z, Dong X. MicroRNA-21 inhibits mitochondria-mediated apoptosis in keloid. Oncotarget. 2017;8(54):92914-92925. doi:10.18632/oncotarget.21656
Burger M, Kaelin S, Leroux JC. The TFAMoplex-Conversion of the Mitochondrial Transcription Factor A into a DNA Transfection Agent. Adv Sci (Weinh). 2022;9(8):e2104987. doi:10.1002/advs.202104987
Mollé LM, Smyth CH, Yuen D, Johnston APR. Nanoparticles for vaccine and gene therapy: Overcoming the barriers to nucleic acid delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2022;14(6):e1809. doi:10.1002/wnan.1809
Xia X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines (Basel). 2021;9(7):734. doi:10.3390/vaccines9070734
Liu Q, Zhang W, Chen S, et al. SELEX tool: a novel and convenient gel-based diffusion method for monitoring of aptamer-target binding. Journal of Biological Engineering. 2020;14(1):1. doi:10.1186/s13036-019-0223-y
Cytochrome c oxidase deficiency: MedlinePlus Genetics. Accessed December 11, 2023. https://medlineplus.gov/genetics/condition/cytochrome-c-oxidase-deficiency/
Bahat A, MacVicar T, Langer T. Metabolism and Innate Immunity Meet at the Mitochondria. Front Cell Dev Biol. 2021;9:720490. doi:10.3389/fcell.2021.720490
Reynolds JC, Lai RW, Woodhead JST, et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat Commun. 2021;12(1):470. doi:10.1038/s41467-020-20790-0
Yong CQY, Tang BL. A Mitochondrial Encoded Messenger at the Nucleus. Cells. 2018;7(8):105. doi:10.3390/cells7080105
Kong BS, Min SH, Lee C, Cho YM. The mitochondrial-encoded MOTS-c prevents pancreatic islet destruction in autoimmune diabetes. Cell Rep. 2021;36(4):109447. doi:10.1016/j.celrep.2021.109447
Gao Y, Wei X, Wei P, et al. MOTS-c Functionally Prevents Metabolic Disorders. Metabolites. 2023;13(1):125. doi:10.3390/metabo13010125
Jiang J, Xu L, Yang L, Liu S, Wang Z. Mitochondrial-Derived Peptide MOTS-c Ameliorates Spared Nerve Injury-Induced Neuropathic Pain in Mice by Inhibiting Microglia Activation and Neuronal Oxidative Damage in the Spinal Cord via the AMPK Pathway. ACS Chem Neurosci. 2023;14(12):2362-2374. doi:10.1021/acschemneuro.3c00140

Tekin S, Bir LS, Avci E, Şenol H, Tekin I, Çınkır U. Comparison of Serum Mitochondrial Open Reading Frame of the 12S rRNA-c (MOTS-c) Levels in Patients With Multiple Sclerosis and Healthy Controls. Cureus. 14(7):e26981. doi:10.7759/cureus.26981

Li S, Wang M, Ma J, et al. MOTS-c and Exercise Restore Cardiac Function by Activating of NRG1-ErbB Signaling in Diabetic Rats. Front Endocrinol (Lausanne). 2022;13:812032. doi:10.3389/fendo.2022.812032
Chen Q, Ruan D, Shi J, Du D, Bian C. The multifaceted roles of natural products in mitochondrial dysfunction. Frontiers in Pharmacology. 2023;14. Accessed December 12, 2023. https://www.frontiersin.org/articles/10.3389/fphar.2023.1093038
Treating the Most Disruptive Symptoms First and Preventing Worsening of Symptoms | Clinical Care of Patients | Healthcare Providers | Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) | CDC. Published April 30, 2021. Accessed December 13, 2023. https://www.cdc.gov/me-cfs/healthcare-providers/clinical-care-patients-mecfs/treating-most-disruptive-symptoms.html
Yao Z, Wan Y, Li B, et al. Berberine induces mitochondrial‑mediated apoptosis and protective autophagy in human malignant pleural mesothelioma NCI‑H2452 cells. Oncology Reports. 2018;40(6):3603-3610. doi:10.3892/or.2018.6757
Cheng Y, Ji Y. Mitochondria-targeting nanomedicine self-assembled from GSH-responsive paclitaxel-ss-berberine conjugate for synergetic cancer treatment with enhanced cytotoxicity. Journal of Controlled Release. 2020;318:38-49. doi:10.1016/j.jconrel.2019.12.011
Can You Overdose on Melatonin? Sleep Foundation. Published May 13, 2021. Accessed December 26, 2023. https://www.sleepfoundation.org/melatonin/melatonin-overdose
Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. Journal of Pineal Research. 2016;61(3):253-278. doi:10.1111/jpi.12360
Tan DX, Manchester LC, Qin L, Reiter RJ. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. International Journal of Molecular Sciences. 2016;17(12):2124. doi:10.3390/ijms17122124

Cecon E, Fernandois D, Renault N, et al. Melatonin drugs inhibit SARS-CoV-2 entry into the brain and virus-induced damage of cerebral small vessels. Cell Mol Life Sci. 2022;79(7):361. doi:10.1007/s00018-022-04390-3
Yu L, Ren L, Dong L. BMAL1 plays a critical role in the protection against cardiac hypertrophy through autophagy in vitro. BMC Cardiovascular Disorders. 2022;22(1):381. doi:10.1186/s12872-022-02822-3
Peng Z, Liang Y, Liu X, Shao J, Hu N, Zhang X. New insights into the mechanisms of diabetic kidney disease: Role of circadian rhythm and Bmal1. Biomedicine & Pharmacotherapy. 2023;166:115422. doi:10.1016/j.biopha.2023.115422
Iweka CA, Seigneur E, Hernandez AL, et al. Myeloid deficiency of the intrinsic clock protein BMAL1 accelerates cognitive aging by disrupting microglial synaptic pruning. Journal of Neuroinflammation. 2023;20(1):48. doi:10.1186/s12974-023-02727-8
Cai H, Liu Y, Men H, Zheng Y. Protective Mechanism of Humanin Against Oxidative Stress in Aging-Related Cardiovascular Diseases. Front Endocrinol (Lausanne). 2021;12:683151. doi:10.3389/fendo.2021.683151
Muzumdar RH, Huffman DM, Atzmon G, et al. Humanin: A Novel Central Regulator of Peripheral Insulin Action. PLoS One. 2009;4(7):e6334. doi:10.1371/journal.pone.0006334
Caricasole A, Bruno V, Cappuccio I, Melchiorri D, Copani A, Nicoletti F. A novel rat gene encoding a Humanin-like peptide endowed with broad neuroprotective activity. FASEB J. 2002;16(10):1331-1333. doi:10.1096/fj.02-0018fje
Saracaloglu A, Mete AÖ, Ucar DF, Demiryürek S, Erbagcı E, Demiryürek AT. Evaluation of Serum Humanin and MOTS-c Peptide Levels in Patients with COVID-19 and Healthy Subjects. Curr Protein Pept Sci. 2023;24(3):277-283. doi:10.2174/1389203724666230217101202
Peluso MJ, Deeks SG, Mustapic M, et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Annals of Neurology. 2022;91(6):772-781. doi:10.1002/ana.26350
Wei DC, Morrison EH. Histology, Astrocytes. In: StatPearls. StatPearls Publishing; 2023. Accessed December 13, 2023. http://www.ncbi.nlm.nih.gov/books/NBK545142/
Ouyang W, Xie T, Fang H, et al. Variable Induction of Pro-Inflammatory Cytokines by Commercial SARS CoV-2 Spike Protein Reagents: Potential Impacts of LPS on In Vitro Modeling and Pathogenic Mechanisms In Vivo. Int J Mol Sci. 2021;22(14):7540. doi:10.3390/ijms22147540
Wassmann S, Stumpf M, Strehlow K, et al. Interleukin-6 Induces Oxidative Stress and Endothelial Dysfunction by Overexpression of the Angiotensin II Type 1 Receptor. Circulation Research. 2004;94(4):534-541. doi:10.1161/01.RES.0000115557.25127.8D
Forcados GE, Muhammad A, Oladipo OO, Makama S, Meseko CA. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front Cell Infect Microbiol. 2021;11:654813. doi:10.3389/fcimb.2021.654813
Patra T, Meyer K, Geerling L, et al. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020;16(12):e1009128. doi:10.1371/journal.ppat.1009128
Forcina L, Miano C, Scicchitano BM, et al. Increased Circulating Levels of Interleukin-6 Affect the Redox Balance in Skeletal Muscle. Oxid Med Cell Longev. 2019;2019:3018584. doi:10.1155/2019/3018584
Hoffman JM, Robinson R, Greenway G, Glass J, Budkin S, Sharma S. Blockade of interleukin-6 trans-signaling prevents mitochondrial dysfunction and cellular senescence in retinal endothelial cells. Exp Eye Res. 2023;237:109721. doi:10.1016/j.exer.2023.109721
Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA Damage and Dysfunction Associated With Oxidative Stress in Failing Hearts After Myocardial Infarction. Circulation Research. 2001;88(5):529-535. doi:10.1161/01.RES.88.5.529
Marasco MR, Conteh AM, Reissaus CA, et al. Interleukin-6 Reduces β-Cell Oxidative Stress by Linking Autophagy With the Antioxidant Response. Diabetes. 2018;67(8):1576-1588. doi:10.2337/db17-1280
Wan W, Zhang L, Lin Y, et al. Mitochondria-derived peptide MOTS-c: effects and mechanisms related to stress, metabolism and aging. J Transl Med. 2023;21:36. doi:10.1186/s12967-023-03885-2
East DA, Fagiani F, Crosby J, et al. PMI: a ΔΨm independent pharmacological regulator of mitophagy. Chem Biol. 2014;21(11):1585-1596. doi:10.1016/j.chembiol.2014.09.019
Parhiz H, Brenner JS, Patel PN, et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J Control Release. 2022;344:50-61. doi:10.1016/j.jconrel.2021.12.027
Hoffman JM, Robinson R, Greenway G, Glass J, Budkin S, Sharma S. Blockade of interleukin-6 trans-signaling prevents mitochondrial dysfunction and cellular senescence in retinal endothelial cells. Exp Eye Res. 2023;237:109721. doi:10.1016/j.exer.2023.109721
Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab. 2013;24(5):222-228. doi:10.1016/j.tem.2013.01.005
Guarnieri J, Dybas J, Fazelinia H, et al. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Science translational medicine. 2023;15:eabq1533. doi:10.1126/scitranslmed.abq1533
Li S, Ma F, Yokota T, et al. Metabolic reprogramming and epigenetic changes of vital organs in SARS-CoV-2–induced systemic toxicity. JCI Insight. 6(2):e145027. doi:10.1172/jci.insight.145027
Zhang L, Richards A, Barrasa MI, Hughes SH, Young RA, Jaenisch R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proceedings of the National Academy of Sciences. 2021;118(21):e2105968118. doi:10.1073/pnas.2105968118
Dhuli K, Medori MC, Micheletti C, et al. Presence of viral spike protein and vaccinal spike protein in the blood serum of patients with long-COVID syndrome. Eur Rev Med Pharmacol Sci. 2023;27(6 Suppl):13-19. doi:10.26355/eurrev_202312_34685
Huynh TV, Rethi L, Lee TW, Higa S, Kao YH, Chen YJ. Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19. Cells. 2023;12(6):877. doi:10.3390/cells12060877
Ripa R, George T, Shumway KR, Sattar Y. Physiology, Cardiac Muscle. In: StatPearls. StatPearls Publishing; 2023. Accessed December 22, 2023. http://www.ncbi.nlm.nih.gov/books/NBK572070/
Pictures Considered #54 : Mitochondria can’t swim... Small Things Considered. Accessed December 22, 2023. https://schaechter.asmblog.org/schaechter/2021/08/pictures-considered-54-mitochondria-cant-swim.html
Du K, Farhood A, Jaeschke H. Mitochondria-Targeted Antioxidant Mito-Tempo Protects against Acetaminophen Hepatotoxicity. Arch Toxicol. 2017;91(2):761-773. doi:10.1007/s00204-016-1692-0
Krauson AJ, Casimero FVC, Siddiquee Z, Stone JR. Duration of SARS-CoV-2 mRNA vaccine persistence and factors associated with cardiac involvement in recently vaccinated patients. NPJ Vaccines. 2023;8:141. doi:10.1038/s41541-023-00742-7
Zhang IW, Curto A, López-Vicario C, et al. Mitochondrial dysfunction governs immunometabolism in leukocytes of patients with acute-on-chronic liver failure. Journal of Hepatology. 2022;76(1):93-106. doi:10.1016/j.jhep.2021.08.009

Mobini S, Chizari M, Mafakher L, Rismani E, Rismani E. Structure-based study of immune receptors as eligible binding targets of coronavirus SARS-CoV-2 spike protein. J Mol Graph Model. 2021;108:107997. doi:10.1016/j.jmgm.2021.107997
Díaz‐Resendiz KJG, Benitez‐Trinidad AB, Covantes‐Rosales CE, et al. Loss of mitochondrial membrane potential (ΔΨ m) in leucocytes as post‐COVID‐19 sequelae. J Leukoc Biol. 2022;112(1):23-29. doi:10.1002/JLB.3MA0322-279RRR

{kind=link}
Dr Kevin McKernan has critiqued the new vaccine integration paper and he doesn't dismiss the paper as fraudulent, but does ask for more confirmations, which isn't surprising at all:
Kevin McKernan
@Kevin_McKernan
As much as I suspect people will eventually find integration, this paper needs a few more confirmations.
1)Same primers were used for nested PCR for 70 cycles.
2)the alignments are noisy.
3)follow up whole genome sequencing or Tag-seq is required to get the flanking genomic seq.
https://x.com/Kevin_McKernan/status/1738083826449236255?s=20
Updated with a new section:
26th December ‘23: (Melatonin: a potent protector of mitochondria).
https://doorlesscarp953.substack.com/p/sars-cov-2-spike-protein-mitochondrial