short story - novelette - novella - novel - PhD thesis - War and Peace - U.S. Tax Code
TL;DR: Safety signals for a plethora of cardiovascular related conditions were being detected from 2020 coincident with SARS-CoV-2 infections. Accounting for a pull forward effect these signals increased again from early 2021 despite deaths attributed to the virus being in decline. Bradford-Hill criteria for “vaccine” associated deaths have been fully met after this period, with excess mortality transitioning to younger age groups up to the most recent data from 2023. Both the virus and synthetic LNP-mRNA platforms distribute systemically and target leucocytes and vascular endothelial linings and plaques, with persistent expression of 6 months to 3 years being indicated. Resultant activation of toll-like receptors, MyD88, cytokine stimulation and upregulation of long-noncoding mRNAs leading to persistent inflammation and proliferation of foam cells is well established in accelerated atherosclerosis and aortic aneurysms. What usually takes decades may take only a few years or even months. It is not unusual to be asymptomatic until the damage is too advanced to be fully reversed. Allopathic medications typically administered for this may fail for the same reason or due to flawed hypotheses'. Where there are benefits these are mostly due to side effects. A new (old?) wholistic approach involving root cause analysis is called for. Mortality could be greatly reduced by instead addressing causative co-morbidities rather than treating symptoms. Alternatives are discussed. Low level inflammation through many of the same pathways discussed contributes to HIV associated cardiovascular disease and is the second largest cause of death of these patients in the US.
Any extracts used in the following article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
On Tuesday 16th January Andrew Bridgen gave an excellent speech at Westminster Hall, London concerning unexplained excess deaths. Its well worth watching:
Although a COVID-19 outbreak in 2020 was associated with excess deaths these were skewed to the elderly and couldn’t explain later increases, which are temporally associated with synthetic gene therapy vaccination programs.
Since 2023 excess deaths have been skewed to younger people, which is extremely disturbing and its excellent that Andrew has been attempting to publicise this scandal.
The first figure includes deaths classified as including COVID-19. The pull-forward effect (PFE) caused by deaths in the older age groups is apparent - you can only die once:
The PFE is evident due to COVID/Midazolam/vents/#3Tablets12 associated homicide in 2020/21:
…Supplies of the sedative midazolam have been diverted from France as a “precaution” to mitigate potential shortages in the NHS caused by COVID-19, the Department of Health and Social Care (DHSC) has told The Pharmaceutical Journal.
…A spokesperson from Accord Healthcare, one of five manufacturers of the drug, told The Pharmaceutical Journal that it had to gain regulatory approval to sell French-labelled supplies of midazolam injection to the NHS, after having already sold two years’ worth of stock to UK wholesalers “at the request of the NHS” in March 2020.
…Midazolam is listed by the Royal College of Anaesthetists as a “first-line” sedative in the management of COVID-19 patients, and warns in guidance published on 2 April 2020 that it “may be subject to demand pressure”.
Matt Hancock, the UK health secretary, told the House of Commons Health and Social Care Select Committee on 17 April 2020 that intensive therapy unit medicines — including midazolam — are part of “a delicate supply chain” as they “are made in a relatively small number of factories around the world”.
From: “Supplies of sedative used for COVID-19 patients diverted from France to avoid potential shortages“ (2020)
But what explains the abrupt snap-back in hospital admissions in 2021/22? Whatever the cause it managed to have an impact great enough to overcome the healthy survivor effect - so much for “95% efficacy and is safe”.3
And as Andrew pointed out, a lack of prescriptions cannot be blamed. Statins don’t reduce net mortality4 and even if they did then then changes in prescription rates would take years to influence cardiovascular death rates and most importantly prescription rates didn’t fall during this period, they actually increased.
Sir Chris Whitty, the chief medical officer, fears thousands missed out on statins and blood pressure medications, resulting in an extra 7,000 non-Covid deaths in the last two years among those aged between 50 and 64. The rise came amid struggles to access GP care since the pandemic, while others were reluctant to burden doctors amid messages to stay home.
From: “Statins should be offered to anyone who wants them, says new NHS guidance
Health service to embark on statins free-for-all, in bid to tackle alarming rise in heart deaths and ease pressure on struggling services“ (2023)
Note that drugs to treat arrythmia also increased significantly over baseline in both 2020 and 2021 (40,331 & 40,591), and hypertensive drug prescriptions also increased over baseline to 73,662 and 73,108, respectively.
In Andrew’s speech he addressed a particular point about Sweden. Although their lock-down rules weren’t as stringent as other countries their vaccination rates were comparable, yet cardiovascular death rates were significantly less than other countries with similar vaccination rates.
How can the vaccines be contributing to these deaths in Sweden, the vaccines must be OK right? Well no, because he emphasises an important point: Sweden was much healthier than other countries to begin with.
If you are physically fit then you are able to tolerate more vaccine poisoning than if you start from a baseline of poor health. There is nothing novel about this principle:
Men who maintained or improved adequate physical fitness were less likely to die from all causes and from cardiovascular disease during follow-up than persistently unfit men. Physicians should encourage unfit men to improve their fitness by starting a physical activity program.
From: “Changes in physical fitness and all-cause mortality. A prospective study of healthy and unhealthy men“ (1995)
Data from the whitewash of a COVID enquiry shows that although there have been repeated waves of infections, these didn’t correlate with deaths recorded as involving coronavirus:
So if its not the virus, why has the ambulance service been under extreme, sustained pressure? What caused the spike in response times at year end ‘22?
At the height of the crisis, it took as long as 9 minutes calls just for the handler to take the call. That must seem like an eternity if, say, your loved one is lying there unconscious.
I don’t recall hearing about this in the MSM at the time, and I don’t recall it ever being investigated:
Doctors are still baffled or blaming everything but the elephant in the room: contaminated experimental gene therapies - toxic LNPs with stabilised synthetic mRNA that persists for months, expressing a cytotoxic oncoprotein in every tissue and organ in your body.
They are either conflicted, wilfully ignorant or plain incompetent.
Nb. these reports aren’t from 2020-21, they are from January 2024:
I like to focus on vax induced pathologies that have tended to receive less attention but are still capable of causing sudden deaths, shortened lifespans and reduced quality of life.
This review presents a hypothesis. Although we have seen that UK deaths recorded as CVD are currently in line with averages from the last few years the LNP/mRNA/spike protein associated signalling pathways invoked are conducive to rapid acceleration of progression of pre-existing plaques.
I won’t be covering all the contributory factors as that could easily fill a book and I’ve covered some in previous Substacks as above. Instead I will focus on pathways that can be traced back to Spike interactions.
The description “accelerated atherosclerosis” is relative. What used to take many decades to develop may instead, for some, become a problem only a few years after transfection.
I would really hope to be wrong on this, but hope isn’t a strategy and research data we already have is a red flag.
Familial hypercholesterolemia is associated with accelerated CAD and deaths of 50% of men by age 50, if untreated7, so this provides a useful benchmark for rapid progression. There is a clinical definition too for timelines measured in months..
What is atherosclerosis? Atherosclerosis is thickening or hardening of the arteries. It is caused by a buildup of plaque in the inner lining of an artery.
Plaque is made up of deposits of fatty substances, cholesterol, cellular waste products, calcium, and fibrin. As it builds up in the arteries, the artery walls become thickened and stiff.
Atherosclerosis is a slow, progressive disease that may start as early as childhood. However, it can progress rapidly.
Its a process that begins in childhood, and the average age of death from CADin the USA was 60 for men and 68 for women.9
Atherosclerosis begins in childhood as an accumulation of fatty streaks-lipid-engorged macrophages (foam cells) and T lymphocytes in the intima of the arteries. Fatty streaks may or may not progress, and may regress. In some people, lipid accumulation is more pronounced with time, and the accumulated lipid becomes covered by a fibromuscular cap to form what is termed a fibrous plaque.
From: “Atherosclerotic Cardiovascular Disease Beginning in Childhood“ (2010)
Autopsies have shown that by our twenties at least half of us show evidence of atherosclerosis.
Further key takes from the above review (emphasis mine):10
Attention was first drawn to the early origin of atherosclerosis by an autopsy study conducted on young soldiers killed in the Korean War.2)Their average age was 22 years, and over 70% of them had evidence of atherosclerosis in their coronary arteries.
Postmortem coronary angiography and dissection of hearts from 105 United States soldiers killed inVietnam demonstrated that 45% had some evidence of atherosclerosis and 5% had gross evidence of severe coronary atherosclerosis.3)
Another study demonstrated a very high incidence of lipid-laden macrophages in the intima of the aorta and coronary arteries of young American children killed in motor accidents, with over 50% of children aged 10-14 years having some evidence of early atherosclerosis.4)
A nation-wide autopsy-based study of atherosclerosis in young Japanese (1 month-39 years) disclosed the presence of fatty streaks in 29% of aortas in those aged <1 year and in 3.1% of coronary arteries of children aged 1-9 years.5) Another examination 13 years later revealed an increased prevalence and extent of coronary artery lesions in autopsied subjects who died in their third and fourth decade of life.6)
In the Bogalusa Heart Study,7) the extent of fatty streaks and fibrous plaques in the aorta and coronary arteries were examined in 204 young patients 2-39-years-of-age. The prevalence of fatty streaks in the coronary arteries increased with age from approximately 50% at 2-15-years-of-age to 85% at 21-39-years-of-age, and the prevalence of raised fibrous-plaque lesions increased with age from 8% at 2-15-years-of-age to 69% at 26-39-years-of-age.
The prevalence and the extent of atherosclerosis was greater with increasing age, body mass index (BMI), blood pressure, and levels of serum total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C). The degree of involvement increased with worsening severity and greater numbers of risk factors.
In the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study,8) the right coronary arteries and aortas were examined during autopsy in 2,876 individuals aged 15-34 years. Raised fatty streaks were present in the abdominal aortas of approximately 20% of those aged 15-19 years and approximately 40% of subjects 30-34-years-of-age, and in the right coronary arteries of approximately 10% of 15-19-year-old subjects and approximately 30% of those aged 30-34 years at the time of death.
The percent of intimal surface involved with raised fatty streaks increased with age and was associated with a high level of non-high-density lipoprotein cholesterol (HDL-C) and low HDL-C, hypertension, obesity, and impaired glucose tolerance.
An intravascular ultrasound study determined that 17% of otherwise healthy heart donors <20 years of age, 37% of those aged 20-29 years, 60% of those aged 30-39 years, 71% of those aged 40-49 years, and 85% of those ≥50-years-of-age had evidence of coronary atherosclerosis.9)
I include an extensive list of preventative therapeutics at the end as most of us are at risk of CVD, especially with advancing years and due to repeated exposure to bio-engineered proteins.
Further to this, a study by López-Melgar et al. (2020) of 3,514 participants (45.7 ± 4.2 years of age; 63% men) using ultrasound found that atherosclerotic plaque build-up occurs rapidly between the ages of 40 and 50, affecting the arteries supplying the heart, brain and legs.
They measured plaque readings and coronary artery calcium scores (CACS) to create a baseline and then again 2.8 years later to record the rate of decline - it can progress quite rapidly. They discovered short term progression of subclinical early atherosclerosis in 41.5% of apparently healthy participants.11
From: “Figure 1. Examples of Atherosclerosis Progression Using Different Imaging Modalities. Intimal irregularities in the carotid bulb (arrow) were detected on visit 1 by 2DVUS. By visit 2, this had progressed to a hypoechogenic atherosclerotic plaque (arrow) extending into the external carotid artery (asterisk). Analysis by 3DVUS detected and quantified significant growth of a plaque already present in the carotid bulb on visit 1 (arrow and asterisk). Computed tomography images show new onset of a calcified lesion in the left anterior descending coronary artery on visit 2 (arrow). 2DVUS = 2-dimensional vascular ultrasound; 3DVUS = 3-dimensional vascular ultrasound; CACS = coronary artery calcium score.” Source: https://www.jacc.org/doi/10.1016/j.jacc.2020.02.026
Endothelial disruption in the arterial wall leads to an aberrant healing immune response. Eventually, usually later in life the plaque may become unstable due to multiple factors and rupture, which can cause sudden arterial blockages (ischaemia) and clot formation (thrombus).12
Foam cells form through dysregulated lipid metabolism in mammalian macrophages: lipid accumulation that exceeds the homeostatic capacity of macrophages triggers lipid droplet formation, which results in the foamy appearance of these macrophages (Box 1). Foam cells are associated with chronic inflammation in certain cancers and in metabolic, infectious, and autoimmune diseases
From: “Foam cells: one size doesn’t fit all“ (2020)
“Macrophage Plasticity and Atherosclerosis Therapy“ (2021) by Lin et al.13 is an excellent review and they discuss how in the earlier stages of plaque formation circulating monocytes are drawn to the site, infiltrate it and phenotypically change to inflammatory type 1 macrophages which then morph into foam cells.
From “FIGURE 1. Main subtypes of macrophages found in atherosclerotic plaques. Distinct stimuli in atherosclerotic microenvironment drive distinct macrophage phenotype differentiation. This figure points out some examples of these macrophage markers or related genes, and the subtypes of macrophages are identified in human (H) and/or mouse (M) atherosclerotic plaques. M1 macrophages can be induced by IFN-γ and LPS, these cells mainly show pro-inflammatory effect. M2 macrophages can be induced by IL-4 and IL-13, and IL-10, these cells can be further divided into M2a, M2b, and M2c subtypes. Of note, these cells are found not always protective.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8138136/
Later, when the plaque is more developed local resident macrophages proliferate within the plaque and lipid-lowering medications cannot stop the progression completely.
Although healing type anti-inflammatory M2 macrophages usually cause healing and plaque regression at a later stage they may lead to plaque instability, rupture and thrombus formation or ischaemia (heart attack).
From “FIGURE 2. Localization of macrophage subtypes in human atherosclerotic lesions. Atherosclerosis is initiated with a local inflammatory response and the presence of monocyte-derived macrophages.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8138136/
In short almost any macrophage infiltration may be associated with cardiovascular pathologies of one sort or another, including microcalcification:
From “FIGURE 3. M1/M2 macrophages and their roles in plaque calcification. Calcification occurs in both the intimal and medial layers of the artery. In the plaque microenvironment, multiple factors, such as inflammation, endoplasmic reticulum (ER) stress, osteoblastic differentiation, hyperlipidemia, and oxidative stress, drive the progression of calcification.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8138136/
Where LNPs & Spike enter the equation is that inflammatory cytokines can lead to proliferation of these same macrophages, and Spike also directly interacts with both CD68+ and CD169+ T cells. Both of these are very common in our immune system and are tissue resident. Pulmonary tissue damage with cytokine storm is another well known pathology caused by COVID-19 Spike binding to these receptors:
Moreover, a transient increase in monocyte-derived macrophage (MDM) levels in lungs was shown by single-cell RNA-seq analysis (13), reflecting the CD68+ cell infiltration that was reported in human COVID-19 cases (23–25). Monocyte infiltration into tissues following viral infections is well described. These cells differentiate and acquire functional macrophage characteristics already in the bloodstream (26) and play a crucial role in shaping the response to infection (27).
From “Spatiotemporal analysis of SARS-CoV-2 infection reveals an expansive wave of monocyte-derived macrophages associated with vascular damage and virus clearance in hamster lungs“ (2023)
Spike associated myocarditis is peculiar in that it also involves CD68+ T cell infiltration:
The results demonstrate a skewed distribution of the number of CD68+ cells in COVID-19 hearts, with upper quantiles showing a significant increase as compared to both matched control hearts, and those with myocarditis. In contrast, hearts from typical inflammatory myocarditis contained increased numbers of CD4+, and CD8+ cells compared to both COVID-19 and control cohorts. In conclusion, the presence of an increased number of CD68+ cells suggests that COVID-19 may incite a form of myocarditis different from typical viral myocarditis, and associated with diffusely infiltrative cells of monocytes/macrophage lineage.
From “COVID-19 myocarditis: quantitative analysis of the inflammatory infiltrate and a proposed mechanism“ (2021)
CD68+ macrophages, if activated in the carotid artery supplying the brain are associated with atherosclerosis, ischaemia and stroke.14 By now you will realise that Spike > CD68+ infiltration leads to a lot of pulmonary and cardiovascular damage.
Virus is not required, just LNPs to circulate the synthetic mRNA everywhere and receptors such as ACE2 or neuropilin-1.
This is a problem as, not coincidentally, ACE2 is expressed on…drumroll… both CD68+ and CD169+ macrophages, leading to upregulation of inflammatory cytokine IL-6.15
Additionally, SARS-CoV-2 viruses infect monocytes which differentiate into tissue macrophages and replicate in their cytoplasm. COVID-19-specific antibodies combine with FcR and enhance virus uptake by macrophages. ACE2 receptor expressed on the surface of lung macrophages interacts with S-protein and allows the SARS-CoV-2 virus to enter the macrophage. S-protein can also interact with ACE2 present on CD68 and CD169 macrophages in the spleen’s marginal area and marginal sinuses of lymph nodes, which upregulates IL-6 production by macrophages.
From: “Immune landscape and redox imbalance during neurological disorders in COVID-19“ (2023)
If this happens in an atherosclerotic plaque it may accelerate progression.
Key takes from “SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels“ (2023) by Eberhardt et al.16 (Emphasis mine):
Patients with coronavirus disease 2019 (COVID-19) present increased risk for ischemic cardiovascular complications up to 1 year after infection.
Although the systemic inflammatory response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection likely contributes to this increased cardiovascular risk, whether SARS-CoV-2 directly infects the coronary vasculature and attendant atherosclerotic plaques remains unknown.
Here we report that SARS-CoV-2 viral RNA is detectable and replicates in coronary lesions taken at autopsy from severe COVID-19 cases. SARS-CoV-2 targeted plaque macrophages and exhibited a stronger tropism for arterial lesions than adjacent perivascular fat, correlating with macrophage infiltration levels.
SARS-CoV-2 entry was increased in cholesterol-loaded primary macrophages and dependent, in part, on neuropilin-1.
SARS-CoV-2 induced a robust inflammatory response in cultured macrophages and human atherosclerotic vascular explants with secretion of cytokines known to trigger cardiovascular events.
Our data establish that SARS-CoV-2 infectscoronary vessels, inducing plaque inflammation that could trigger acute cardiovascular complications and increase the long-term cardiovascular risk.
VSMCs: Vascular smooth muscle cells.
To further investigate SARS-CoV-2 infection of VSMCs and lipid-laden VSMCs, which are associated with atherosclerosis17–20, we infected primary human aortic VSMCs, and VSMCs loaded with cyclodextrin-cholesterol complexes (Extended Data Figure 2d), with the SARS-CoV-2 USA WA1/2020 isolate.
Approximately 18% of cultured VSMCs and 13% of cholesterol-loaded VSMCs were spike+, and the frequency of spike antisense+ACTA2+ cells, indicating viral replication, was ≈2.6% (Extended Data Figure 2d–e).
Taken together with our in vitro findings, which indicate that more than 79% of macrophages and over 90% of foam cells are spike+, along with the discovery that more than 40% of both cell types are spike antisense+, these results show that although SARS-CoV-2 can infect vascular smooth muscle cells (VSMCs), macrophages are infected at a higher rate.
Why these “complications” should be resolved in one year needs following up, as dead myocardium cannot just regenerate. It becomes fibrotic and the heart wall thickens and works harder to compensate (remodelling).
Atherosclerotic lesions which have progressed during this period are unlikely to regress back to the starting condition, at least without strong meds throughout as a prophylactic.
When you look into their source reference from 2022 it didn’t show recovery at 12 months, they actually lacked data beyond this and used projections.17
What does stand out is that the range of cardiovascular pathologies from their figure correlates well with vax induced incidents from 21/22, post COVID-19.
It reads like a summary of VAERS reports:
From: “Fig.2.Risks and 12-month burdens of incident post-acute COVID-19 cardiovascular outcomes compared with the contemporary control cohort. Outcomes were ascertained 30 d after the COVID-19-positive test until the end of follow-up. COVID-19 cohort (n = 153,760) and contemporary control cohort (n = 5,637,647). Adjusted HRs and 95% CIs are presented. The length of the bar represents the excess burden per 1,000 persons at 12 months, and associated 95% CIs are also shown.”, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8938267/
Rapid progression of atherosclerosis
Prior to 2020, accelerated atherosclerosis was observed in a limited number of patients with known risk factors or medical histories. In 2015 Shah et al. discussed some of these factors and offer a definition for rapid progression.
Key takes from “Rapid Progression of Coronary Atherosclerosis: A Review“18 (emphasis mine):
Vascular injury is believed to be critical initiating event in pathogenesis of spontaneous atherosclerosis. Syndrome of accelerated atherosclerosis has been classically described in patients undergoing heart transplantation, coronary artery bypass graft, and percutaneous transluminal coronary angioplasty.
In contrast to spontaneous atherosclerosis, denuding endothelial injury followed by thrombus formation and initial predominant smooth muscle cell proliferation is believed to be playing a significant role in accelerated atherosclerosis.
There is no universal definition of rapid progression of atherosclerosis. However most studies describing the phenomenon have used the following definition: (i) > or = 10% diameter reduction of at least one preexisting stenosis > or = 50%, (ii) > or = 30% diameter reduction of a preexisting stenosis <50%, and (iii) progression of a lesion to total occlusion within few months.
Recent studies have described the role of coronary vasospasm, human immunodeficiency virus, various inflammatory markers, and some genetic mutations as predictors of rapid progression of atherosclerosis. As research in the field of vascular biology continues, more factors are likely to be implicated in the pathogenesis of rapid progression of atherosclerosis.
Although atherosclerosis is believed to progress over many years, it has been increasingly noted to progress over few months to 2-3 years in few patients without traditional factors for accelerated atherosclerosis. Hence the term rapid progression of atherosclerosis has been used in recent years.
In spontaneous atherosclerosis, there is chronic damage to arterial endothelium by turbulence of blood flow or other injuries, which leads to nondenuding functional alterations of endothelial cells (type 1 injury). This leads to lipid accumulation, the initial predominant feature in this type. Adhesion of monocytes and platelets either simultaneously or at a later time occurs. Along with altered endothelium, these cells release various growth factors, leading to migration and proliferation of smooth muscle cells. This ultimately forms a typical atherosclerotic plaque [2].
In contrast to spontaneous atherosclerosis, accelerated atherosclerosis is initiated by significant denuding endothelial injury (type 2 or 3 injury). Once endothelium is denuded, immediate platelet aggregation and thrombus formation occur on subendothelium. Intact endothelium is a potent inhibitor of growth of smooth muscle cells. Hence endothelial denudation leads to early smooth muscle cell proliferation and fibrosis mediated by various factors released by platelets, leucocytes, and smooth muscle themselves. Accumulation of lipids occurs late in accelerated atherosclerosis [2].
Just published in “Trends in Microbiology” at time of writing and Perico et al. review how the virus and Spike alone can induce severe endothelial damage, including by denuding vessels of their glycocalyx (GCX). Loss of GCX volume is associated with accelerated atherosclerosis in type 2 diabetic patients.19
Key takes from: “SARS-CoV-2 and the spike protein in endotheliopathy“20 (2023, emphasis mine):
Endothelial cells are located in the inner layer of blood vessels, forming a continuous monolayer that functions as a barrier [11.]. Endothelial cells and adjacent pericytes contribute to preserving vascular hemostasis by maintaining vessel integrity, balancing fibrinolysis through the expression of coagulation inhibitors and blood clot-lytic enzymes, and maintaining the glycocalyx [12.].
A large body of evidence is growing that shows that critically ill COVID-19 patients exhibit severe glycocalyx damage [49.]. The glycocalyx is a key regulator of endothelial cell homeostasis and fulfills diverse functions, ranging from mechanotransduction and the maintenance of vascular integrity and vascular tone [50.].
Impaired endothelial glycocalyx has been associated with vascular dysfunction in convalescent COVID-19 patients 4 months after infection [56.]. Signs of severe endothelial glycocalyx deterioration are observed in children with multisystem inflammatory syndrome (MIS-C) related to COVID-19 [57.]. Based on the above, preventing endothelial glycocalyx degradation could be an attractive therapy for COVID-19 [58.].
The first insights into the mechanism through which the spike protein impacts endothelial glycocalyx was provided by Biering and colleagues who documented that the spike protein engages GAGs on the cell surface via positively charged surfaces in the RBD (Figure 3) [59.].
The spike protein bound to GAGs can interact with different integrins, which mediate SARS-CoV-2 infection in an ACE2-independent manner via endocytic trafficking and autophagy [60., 61., 62.]. The spike protein binding to integrins also induces the activation of the transforming growth factor (TGF)-β pathway, which, in turn, modulates key enzymes involved in glycocalyx regulation, such as heparanase, hyaluronidases, and neuraminidase, leading to the disruption of intercellular junctional complexes and vascular barrier dysfunction (Figure 3) [59.].
From: “Figure 3. The spike protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) engages intracellular pathways involved in glycocalyx degradation and cytokine production in endothelial cells.” Source: https://www.cell.com/trends/microbiology/fulltext/S0966-842X(23)00189-0
The potential role of the spike protein in vaccine-induced adverse events
…mRNA-based vaccines encode a recombinant membrane-bound spike with specific amino acid variations introduced to maintain the protein in a prefusion state and uncleavable form [107.,108.]. Due to these modifications, the systemic bioavailability of spike protein or its subunits was initially excluded until a study reported that vaccine mRNA can be detected in blood at 15 days post-vaccination, suggesting a potential systemic delivery of the lipid nanoparticles containing the synthetic spike mRNA [109.].
“Likely” means an unsubstantiated opinion. Case reports on a large scale render this a mute point. It may be low because it is not in circulation. Clinical data and other studies usurp these comments:
Consistently, circulating S1 subunit can be detected 1 day after the first injection of mRNA-1273 COVID-19 vaccine, up to 150 pg/ml and for about 2 weeks after injection [110.]. These amounts of circulating S1 in the range of pg/ml are likely too low to trigger endothelial cell dysfunction, while higher levels, in the range of ng/ml, are required in mice to mimic the endotheliopathy observed in severe COVID-19 cases [59.]. In keeping with this, circulating S1 was detected in severe COVID-19 patients with maximum levels of about 1 ng/ml and higher S1 levels correlated with ICU admission [111.].
The suggestion that the spike protein generated following mRNA vaccination could have a limited impact on endothelial cell function was provided by data from 32 vaccine recipients, which showed that that mRNA vaccine caused only a moderate, transient deterioration of endothelial function, which returned to normal levels after 2 days from vaccination [112.].
More recently, the circulating spike protein has been implicated in extremely rare cases of cardiovascular complication following mRNA COVID-19 vaccination in young males [113.].
Indeed, a study showed the presence of high levels of full-length unbound spike protein that remained detectable for up to 3 weeks after vaccination in adolescents who developed post-vaccine myocarditis, compared with the asymptomatic cohort [114.].
Notably, the circulating spike protein eluded antibody recognition [114.]. Extensive antibody profiling and T cell responses in the individuals who developed postvaccine myocarditis were essentially indistinguishable from those of vaccinated control subjects [114.], possibly advancing the hypothesis that circulating spike protein is the underlying cause of post-vaccine myocarditis in this cohort.
Its 2024 and we still don’t have any more biodist information. This is revealing in itself, how long do we have to wait to confirm the damage mechanisms?
While the benefit–risk profile continues to favor COVID-19 vaccination, these studies suggest that an age difference in the processing and clearance of the spike protein translated from the mRNA vaccine can occur. The biodistribution of COVID-19 vaccines has not been systematically analyzed in humans, and future research is warranted to understand the localization, synthesis, and disposal of vaccine-induced spike protein.
4 ways that Spike can penetrate vascular endothelium and leukocytes
1/ Via lipid nanoparticles (LNPs) & transfection
The LNPs they used are pegylated in order to reduce rates of phagocytosis by macrophages, but they can still be endocytosed by hepatic macrophages due to their lipoprotein receptors.21 This is one of the reasons why LNPs accumulate in the liver and cause toxicity.22
In recent years, extensive studies have revealed that, surface functionalization, in addition to size, shape and mechanical rigidity, is a key factor that influences the phagocytic fate of particles.6–8Engineering the surface chemistry of particles to prevent their internalization by immune cells has been the focus of many studies.9–14 One widely used strategy is to coat particles with a layer of polyethylene glycol (PEG).15–17 As particles move in the blood stream, opsonins such as antibodies and serum proteins adsorb on the particles and “mark” them for phagocytic removal. A dense coating of the hydrophilic PEG shields synthetic particles from macrophage internalization by creating a hydration layer that prevents the nonspecific adsorption of opsonins onto particles and reduces cell adhesion.18–22
from: “Effect of partial PEGylation on particle uptake by macrophages“ (2016)
Dendritic cells (DCs), macrophages and monocytes can be directly transfected, although B cell activation is the principle mechanism.
Key takes from “mRNA vaccines for COVID-19: what, why and how“ by Woo et al.23 (2021, emphasis mine):
1. mRNA-1273: It was loaded into two proprietary cationic LNPs, WO2017070626 and WO2018115527. Although the exact formulation is not known, the composition of the LNPs was described as follows, SM-102, polyethylene glycol-2000-dimyristoyl glycerol (PEG2000-DMG), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) 80.
2. BNT162b mRNA: It was encapsulated by patented LNPs with improved efficiency of the mRNA delivery according to its clinical trial report (#NCT04368728) 81, 82. The LNPs are composed of ionizable amino lipid, phospholipid, cholesterol and a PEGylated lipid prepared at a ratio of 50:10:38.5:1.5 mol/mol 82, 83. It is interesting to note that BTN162b and mRNA-1273 vaccines are suggested to be shipped and stored at -80˚C and -20°C, respectively 80, 82.
…the majority of the protein will be degraded in endosome-derived proteasome and subsequently incorporated as a part of the class I major histocompatibility complexes (MHCs), and presented to CD8+ and CD4+ T cells, respectively 53,59. Dendritic cells transfected by an mRNA vaccine or its endocytosed immunogens process the assembly of the class II MHC complex and present it to immune cells(Figure 2).
From: “Figure 2. Delivery and working mechanism of a mRNA vaccine. mRNA vaccine, containing the coding region of S protein flanked by the optimized 5'- and 3'-UTRs and polyA tail, is synthesized via IVT, followed by 5'-capping with a 5'-cap analogy and encapsulation with LNP for IM injection (step 1). The vaccine is delivered into muscle cells or antigen-presenting cells such as dendritic cells or macrophages via endocytosis (step 2).“, source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8071766/
Papers by Pradhan et al. and others discussed the GAG and V3 variable loops with homology to gp120 from HIV that couldn’t get there by chance or zoonotic transfer into the bioweapon that became SARS-CoV-2.24 Researchers started decades ago using serial passage through humanised mice and gain of function techniques to create variants of chimpanzee (simian) SIV viruses that could invade and proliferate in human tissues under the new name of HIV.25
CRISPR-Cas9 type gene editing and splicing techniques were then later used to weaponize SARS-CoV-2.
Nb. None of this is tinfoil hat conspiracy stuff. Work by Baric, Sirotkin, Schmitt etc is no secret, they just would rather you memory hole it and blame bats, pangolins and racoon dogs instead:
I don’t buy the narrative that safe vaccines were the objective as these never worked as sold, yet we always seem to get the novel of GOF enhanced pathogen from it such as RSV, influenza, hepatitis, Lyme’s.
Quite a lot of work went into getting HIV epitopes into human coronaviruses, under the guise of “vaccine” development.
This is one of the reasons gp120 is so useful, and it is all relevant to synthetic mRNA mediated accelerated-atherosclerosis:
…DC-SIGN is a C-type lectin that binds to endogenous adhesion molecules ICAM-2 and ICAM-3 as well as the viral envelope glycoprotein human immunodeficiency virus, type 1, glycoprotein (gp) 120. We wished to determine whether DC-SIGN binds differently to its endogenous ligands ICAM-2 and ICAM-3 versus HIV-1 gp120. We found that recombinant soluble DC-SIGN bound to gp120-Fc more than 100- and 50-fold better than ICAM-2-Fc and ICAM-3-Fc, respectively. This relative difference was maintained using DC-SIGN expressed on three different CD4-negative cell lines.
From: “DC-SIGN Binds to HIV-1 Glycoprotein 120 in a Distinct but Overlapping Fashion Compared with ICAM-2 and ICAM-3“ (2004)
…First, the receptor of HCoV 229E, human aminopeptidase N (hAPN or CD13) is expressed mainly on human dendritic cells (DCs) and macrophages indicating that targeting of HCoV 229E-based vectors to professional antigen presenting cells can be achieved by receptor-mediated transduction. Second, HCoV 229E structural genes can be replaced by multiple transcriptional units encoding various antigens. These virus-like particles (VLPs) containing HCoV 229E-based vector RNA have the ability to transduce human DCs and to mediate heterologous gene expression in these cells.
…In contrast to viral vectors based on DNA viruses, the use of positive-stranded RNA virus-based vectors that replicate in the cytoplasm are considered as safe vectors because it is unlikely that sequences from these vectors can integrate into the host cell genome.
…The primary goal of the outlined approach is the establishment of the coronavirus vector system and its validation in a small animal model. If this approach is feasible and effective, we should commence with the development of HCoV 229E replicon-based VLPs encoding several HIV antigens (env, gag and nef) in combination with immunostimulatory molecules. The successfully established packaging strategy will be adapted to the HCoV 229E system and should allow production of recombinant HCoV 229E VLPs. Alternatively, pseudotyped MHV-based VLPs displaying a tropism for human DCs may be used for further studies. Safety and efficacy of this vaccine preparation should be tested in an adequate non-human primate model
From: “Towards a Coronavirus-Based HIV Multigene Vaccine“ (2006)
This is from his CV, which is quite publicly posted on multiple websites but I won’t link to it as it includes a “home address” and I’m not into accusations of “doxxing”:
National Institutes of Health, Allergy and Infectious Diseases. R21/R33 AI 076159-03 Human Coronaviruses as Multigene Mucosal Vaccine Vectors for HIV (Sims-PI; Baric Co-Investigator); Total Direct costs: $286,661. 04/01/08 - 03/31/11
This project will provide the first critical evaluation of the potential use of common cold human coronaviruses as live mucosal vaccine vectors for HIV.
2/ Viafree Spike in the blood
An association has been found between elevated levels of full length Spike in plasma and myocarditis, as touched on briefly earlier.
Key takes from “Circulating Spike Protein Detected in Post–COVID-19 mRNA Vaccine Myocarditis“ by Yonker et al.27 (2023, emphasis mine):
Extensive antibody profiling and T-cell responses in the individuals who developed postvaccine myocarditis were essentially indistinguishable from those of vaccinated control subjects, despite a modest increase in cytokine production.
A notable finding was that markedly elevated levels of full-length spike protein (33.9±22.4 pg/mL), unbound by antibodies, were detected in the plasma of individuals with postvaccine myocarditis, whereas no free spike was detected in asymptomatic vaccinated control subjects (unpaired t test; P<0.0001).
Immunoprofiling of vaccinated adolescents and young adults revealed that the mRNA vaccine–induced immune responses did not differ between individuals who developed myocarditis and individuals who did not. However, free spike antigen was detected in the blood of adolescents and young adults who developed post-mRNA vaccine myocarditis, advancing insight into its potential underlying cause.
Cleaved Spike S1 is persistent for weeks but rarely associated with myocarditis:
When analyzed according to time since vaccination, free S1, which was detected in only one patient with postvaccine myocarditis and one vaccinated control subject, was detected only within the first week. However, antibody-bound S1, which was detected in roughly one-third of both cohorts, could be detected up to 3 weeks after vaccination (Figure (Figure4B).4B). In contrast, both free and antibody-bound spike, which was detectable only in patients who developed vaccine-induced myocarditis, remained detectable up to 3 weeks after vaccination (Figure (Figure4B).4B).
Plausible mechanisms. The irony here is that a lack of Spike clearing immune response is implicated.
There is growing in vitro evidence that spike itself can stimulate cardiac pericytes dysfunction23 or inflame the endothelium, potentially by downregulating angiotensin-converting enzyme 2 expression, by impairing endothelial nitric oxide bioavailability,24 or by activating integrin-mediated inflammation with hyperpermeability of the endothelial cell layer.25Thus, the spike antigen itself, which evades antibody recognition rather than invoking immune hyperactivation, may contribute to myocarditis in these individuals.
Hello, Spike tolerance and igG4 class switching anyone?
Unfortunately they didn’t assay this but did measure igM, igA1 & igG1.
It has previously been shown that after the first inoculation of the mRNA-1273 vaccine, the cleaved S1 subunit of spike can be detected in the plasma of healthy adults.16 However, after the second dose, no antigen was detected,16 presumably because there are higher levels of circulating anti–SARS-CoV-2 antibodies, which quickly bind any circulating antigen, facilitating its clearance.
In contrast, one-third of the adolescents displayed antibody-bound S1 antigenemia after the second vaccination, regardless of the development of myocarditis, a finding not seen in our smaller sample of adults. This suggests that either the immune system of adults responds more quickly to the vaccine-induced production of spike or, because of differences in body mass, the levels of S1 fall below the limit of detection for adults.
Alternatively, increased levels of free spike compared with free S1 may be attributable to differences in renal clearance rates; S1 would be expected to clear faster with a molecular weight of 76 kDa, approximately half that of full spike (180 kDa). Because both adults and the adolescents included in our cohort received adult dosing of the mRNA vaccine, this finding suggests an age-related capacity for handling vaccine-introduced antigen.
It is important to note that the majority of circulating S1 was bound by specific anti-S1 antibodies, indicating an appropriate immune response for targeting and clearing S1.
3/ Viaexosomes expressing Spike
Exosomes are derived from endosomes. Milosevits et al. (2015) discussed why exosomes can make great delivery vehicles, the key point being that they are immune privileged.
Key takes from ”Exosomes: potential model for complement-stealth delivery systems”28 (emphasis mine):
Exosomes were first described in the 1980s by Johnstone et al. (5) and were defined as vesicles formed in the endosomal compartments (multivesicular endosomes) which then get secreted into the extracellular space (Figure 1) to serve as nano-rafts carrying biological information between cells. Hence, they play a central role in intercellular communication (5).
From “Figure 2: Electron micrographs of dendritic cell derived exosomes and microvesicles. The size bars in the EM images indicate 100 nm. The images were reproduced with permission from (19).” Source: https://www.degruyter.com/document/doi/10.1515/ejnm-2015-0005/html
The proteins attached to the lipid bilayer of exosomes (arrows) originate from the plasma membrane which is preserved its original orientation. They cover a broad spectrum of immune-modulating and cell recognizing molecules that are either common, ubiquitous proteins or cell-type specific proteins.
…the surface exposed proteins have different roles including the targeting of exosomes to specific cells and modulating the immune response via activation or suppression. Additionally it has to be mentioned that the fact that endogenous exosomes are made from fragments of the plasma membrane in the preserved original orientation means that they also inherit the glycome, the glycocalyx from the originating cells with its innate immune tolerance.
That exosome loading with drug molecules or therapeutic proteins, peptides, and hormones is feasibleis clearly indicated by the negative example of the pathogenic exosome-loading of prion (89), or other disease-related proteins (90).
Key takes from “Cutting Edge: Circulating Exosomes with COVID Spike Protein Are Induced by BNT162b2 (Pfizer-BioNTech) Vaccination prior to Development of Antibodies: A Novel Mechanism for Immune Activation by mRNA Vaccines” by Bansal et al.29 (2021, emphasis mine):
To determine the mechanism, we analyzed the kinetics of induction of circulating exosomes with SARS-CoV-2 spike protein and Ab following vaccination of healthy individuals. Results demonstrated induction of circulating exosomes expressing spike protein on day 14 after vaccination followed by Abs 14 d after the second dose.
Exosomes with spike protein, Abs to SARS-CoV-2 spike, and T cells secreting IFN-γ and TNF-α increased following the booster dose.
This stuff is circulating systemically for 4 months after transfection. It doesn’t exactly “stay in the arm”.
Transmission electron microscopy of exosomes also demonstrated spike protein Ags on their surface. Exosomes with spike protein and Abs decreased in parallel after four months.
These results demonstrate an important role of circulating exosomes with spike protein for effective immunization following mRNA-based vaccination.This is further documented by induction of humoral and cellular immune responses in mice immunized with exosomes carrying spike protein.
This is what the exosomes look like, with the Spike presenting on the surface:
From: “FIGURE 1. (A) Representative NanoSight image for exosomes from vaccinated individuals with mean and median sizes (black thin line in the graph indicates the three measurements of the same sample, and red line is the average of all three lines). (B) Transmission electron microscopy images of SARS-CoV-2 spike Ag on exosomes from control exosomes from control and vaccinated individuals. Arrows indicate SARS-CoV-2 spike-positive exosomes. Right side, third image is the zoomed image of positive exosome from vaccinated sample (original magnification x 50,000). We have used anti-coronavirus FIPV3-70 Ab as negative control for both the samples.” Source: https://journals.aai.org/view-large/figure/8250243/ji2100637f1.tif
Extract from: “FIGURE 2…(D) Before and after line plot of levels of SARS-CoV-2 spike Ab in vaccinated healthy individuals at day 14-1, day 14-2, and 4 mo after second dose of vaccine (E) Before and after line plot of densitometry of Western blots for SARS-CoV-2 spike protein in vaccinated healthy individuals at day 14-1, day 14-2, and 4 mo after second dose of vaccine.” Source: https://journals.aai.org/view-large/figure/8250248/ji2100637f2.tif
4/ Viaexosomes containing synthetic mRNA
In 2023 Hanna et al. conducted investigations into whether synthetic mRNA was detectable in breast milk after vaccination and whether it was potentially capable of expressing Spike?
Short answer is that, in this case, it was detectable but in what they describe as “trace mRNA amounts”, highly degraded and incapable of expressing proteins.
Key takes from “Biodistribution of mRNA COVID-19 vaccines in human breast milk“30 (emphasis mine):
Here, we evaluated if COVID-19 vaccine mRNA is detectable in BM after maternal vaccination and determined its potential translational activity.
We collected BM samples from 13 lactating, healthy, post-partum women before and after COVID-19 mRNA vaccination. Vaccine mRNA in whole BM and BM extracellular vesicles (EVs) was assayed using quantitative Droplet Digital PCR, and its integrity and translational activity were evaluated.
Of 13 lactating women receiving the vaccine (20 exposures), trace mRNA amounts were detected in 10 exposures up to 45 h post-vaccination. The mRNA was concentrated in the BM EVs; however, these EVs neither expressed SARS-COV-2 spike protein nor induced its expression in the HT-29 cell line. Linkage analysis suggests vaccine mRNA integrity was reduced to 12–25% in BM.
A further complication is that the synthetic mRNA carrying LNPs themselves may be trafficked from cell to cell in exosomes:
Upon internalization, mRNA LNPs are routed through the endo-lysosomal compartment, where most of the mRNA LNPs remain entrapped in endosomes and degrade over time. The intracellular trafficking and underlying mechanisms on how LNPs enable the escape of mRNA from the endosomes to reach the cytoplasm are still not fully understood [[43]
From: “The dawn of mRNA vaccines: The COVID-19 case“ (2021)
What we can take from these 4-5 carrier mechanisms is that PEGylation is a mute point regarding prevention of phagocytosis by macrophages. The Spike will get in there regardless - it can distribute to almost any tissue via the exosomal trafficking.
COVID-19 and atherosclerosis
Key takes from “Atherosclerosis, Cardiovascular Disease, and COVID-19: A Narrative Review” by Vilaplana-Carnerero et al.31 (2023, emphasis mine).
With this statement a whole multi-billion dollar industry is exposed as a scam for most. Blood pressure (BP) control through anti-hypertensive drugs without addressing underlying causes is largely just treating a symptom:
Older patients with controlled blood pressure diagnosed for longer had a higher risk of death by COVID-19 than those with a recent diagnosis of hypertension and uncontrolled blood pressure. The authors hypothesized that older individuals could have more advanced atherosclerosis and target organ damage that predispose to worse COVID-19 outcomes [41].
Although a review from 2023, vaccination status is not disclosed.
Spike mediated cardiovascular pathologies are likely to be much lower in the unvaccinated and through natural infection due to the rapid interception of causative viruses and free Spike through activation of the complement system, T-cell immunity and a range of neutralising antibodies including mucosal igM, gut igA and serum igG1 & igG332.
...On the other hand, the study of Pillarisetti et al. analyzed data of the TriNetX COVID-19 global research network, a cohort of 81,844 patients with a diagnosis of COVID-19. The study found that 9.3% of patients developed cardiac complications after diagnosis of COVID-19. Heart failure, atrial fibrillation, sinus bradycardia, and acute coronary syndrome were the most incident. Death occurred in 20% of patients with cardiac complications. Although the analysis was adjusted for sex and greater number of comorbidities, the mortality was significantly higher in that group than in the one without cardiac complications [49].
...Long COVID is defined as the continuation or development of new symptoms three months after the initial SARS-CoV-2 infection, with these symptoms lasting for at least 2 months with no other explanation [50]
Currently, three years after the declaration of the pandemic, emerging long-term outcome data demonstrate a significant burden of CVD following acute infection with SARS-CoV-2 [51].
Informative comments from “Vigilance on New-Onset Atherosclerosis Following SARS-CoV-2 Infection” (2021) by Liu et al.33 (emphasis mine):
Endothelial dysfunction is an initial step in the development of atherosclerosis that precedes clinical symptoms and has prognostic value for future cardiovascular events (34, 35).
Furthermore, endothelial dysfunction emerges as one of the essential mechanisms corresponding to the enhanced atherosclerotic risk among HIV, HCV and other viral infected people (14, 36, 37).
Therefore, endothelial dysfunction induced by SARS-CoV-2 infection indeed becomes a strong contributor to upcoming atherosclerosis in subjects who have recovered from COVID-19.
From “Figure 1. Role of SARS-CoV-2 in endothelial dysregulation. Endothelial dysfunction is an initial step in the development of atherosclerosis that precedes clinical symptoms. SARS-CoV-2 can induce endothelial damage directly or indirectly by eliciting immune dysregulation which causes cytokine storm, leading to the deteriorations of endothelial damage.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7855580/
SARS-CoV-2 has been found to efficiently infect immune cells expressing low ACE2, such as macrophages and T lymphocytes, through CD147-mediated viral entry (55). Therefore, CD147 is upregulated and possibly participates in hyperinflammation induced by SARS-CoV-2. Accumulating studies have highlighted the potential proatherosclerotic effects of CD147 in atherosclerosis (56).
Bonus pathology: Aortic dissection
For background, Badaras et al. go into further detail of how SARS_CoV-2 can lead to endothelial damage. Of critical importance Spike alone can mediate this damage and cause both accelerated atherosclerosis and aortic aneurysms & dissection through common mechanisms.
Aortic aneurysm: A balloon like bulge in the aorta that can dissect or rupture.
Aortic dissection: A tear in the inner layer of the the body’s main artery, the aorta.
“Diagram of aortic aneurysm Figure A shows a normal aorta. Figure B shows a thoracic aortic aneurysm (which is located behind the heart). Figure C shows an abdominal aortic aneurysm located below the arteries that supply blood to the kidneys.” Source (public domain): http://www.nhlbi.nih.gov/health/health-topics/topics/arm/types.html
I’m hearing more and more cases each week of sudden deaths in middle age (40’s + usually) due to aortic dissection in otherwise previously “healthy” asymptomatic, but multiple boosted adults.
Key takes from “Vascular Aging and COVID-19”34 (2022, emphasis mine):
The damage SARS-CoV-2 does to the endothelial wall can be split into two parts: direct and indirect damage. Indirect damage is caused by hyper inflammation and an increase in circulating cytokine levels.81
cfPWV:Carotid‐femoral pulse wave velocity. There is an association between cfPWV, arterial stiffness, CVD and pathogenesis of hypertension.35
CRP: C-reactive protein is a protein made by the liver in response to Il-6 secretion by macrophages and is biomarker for systemic inflamation:36
Previous studies have shown that acute infection can result in increased cfPWV, possibly by decreasing NO bioavailability.82In vivo studies have shown that C-reactive protein reduces eNOS expression and activity in endothelial cells, thus leading to functional stiffening of the arteries.83
cfPWV increase during acute infection also strongly correlates with C-reactive protein, IL-6 and matrix metalloproteinase-9 (MMP-9) levels.84 The increase in MMP-9 levels leads to reduced elastin synthesis and fragmentation.85
Healthy elastin prevents vascular smooth muscle cells from changing their phenotype from normal contractile phenotype to pathologic secretory phenotype.86Increased arterial stiffness leads to increased damage to the arterial wall due to changes in pulse pressure.87
This arterial damage itself leads to atherosclerosis and inflammation, and these effects both contribute to arterial stiffening.88,89 Thus, a vicious cycle is formed.1 Increased arterial stiffness has been proven to cause target organ damage, it is also used to predict CV events and mortality. Infection of the endothelial cell leads to endothelial dysfunction through impaired smooth muscle cell function and vascular extracellular matrix remodelling.83
Spike protein alone (ie from synthetic mRNA) is sufficient to induce synthesis of factors crucial for atherosclerosis to develop and progress:
VCAM-1, ICAM-1 and various pro-inflammatory cytokines such as tumour necrosis factor alpha (TNFa), Il-1 and IL-6, as above.
MMP-9 has been shown to promote the formation of new atherosclerotic plaques and instability of plaques.96,97In vitro studies have demonstrated that SARS-CoV-2 spike protein is enough to induce the synthesis of adhesion molecules (VCAM-1 and ICAM-1) and pro-inflammatory cytokines (TNFα, IL-1β and IL-6) in human umbilical vein endothelial cells.98Pro-inflammatory cytokines contribute to the progression of atherosclerosis, and the expression of adhesion molecules is crucial for the development of atherosclerosis.99,100 This shows a reasonable theoretical pathway of SARS-CoV-2-induced atherosclerosis.
And the link to aortic aneurysm...
A study from 2010 by Fan et al. used rats, and the clues are in the title. Vascular inflammation is strongly associated with abdominal aortic aneurysm progression.
Key takes from “MCP-1, ICAM-1 and VCAM-1 are present in early aneurysmal dilatation in experimental rats”37 (emphasis mine):
Recent studies have suggested that inflammation actively participates in ascending aortic aneurysm formation. The aim of the present study was to evaluate the expression changes of adhesion molecules and MMPs in an experimental model of ascending aortic aneurysm induced by ascending aorta banding in Wistar rats.
Twelve rats developed aortic dilation after ascending aorta banding treatment, while nine normal animals underwent surgery without banding were used as controls. Light microscope and scanning electron microscope showed that the wall of the ascending aorta became disorganized as well as infiltration by inflammatory cells in aneurysmal rats.
MCP1: Monocyte chemoattractant protein-1, a CC chemokine that strongly attracts leukocytes as part of an inflammatory response caused by infection, damage or localised Spike: “The mRNA and protein expression levels of the inflammatory cytokines, IL-6, TNF-α, IL-1β, and IFN-γ, and the chemokine,MCP-1, were markedly increased by overexpression of SARS-CoV-2 spike protein”.38
By using immunohistochemical techniques, a significant increase in the immunostaining of MCP-1 was observed in the aneurysmal wall as compared to the normal aortic wall. Under similar experimental conditions, we also found that the immunostaining of ICAM-1 and VCAM-1 was markedly increased in the aneurysmal wall.
MMP-9: Matrix Metalloproteinase-9 is involved with pathological remodelling processes such as fibrosis, inflammation and, most critically, the degradation of proteins that maintain the integrity of endothelial barriers - ECM proteins or extracellular matrix.39
“Interestingly, it was found that MMP-9 and ferritin are the key mediators along with other cytokines like IL-6, IL-10, IL-17, TNF-α, and IL-1β during the S-protein mediated signaling.”40
In addition, gelatin zymographic analysis showed that the expression and activities of MMP-2 and MMP-9 were remarkably enhanced in the ascending aorta of ascending aortic aneurysmal rats as compared to normal rats.
These results demonstrate that MCP-1, ICAM-1 and VCAM-1 are involved in the pathogenesis of ascending aortic aneurysm and an increase in the immunostaining and activity of MMP-2 and MMP-9 may promote the progression of ascending aortic aneurysm.
In human sporadic AAD tissues, we observed the presence of cytosolic DNA in SMCs and macrophages and significant activation of the STING pathway. In the sporadic AAD model, Stinggt/gt mice showed significant reductions in challenge-induced aortic enlargement, dissection, and rupture in both the thoracic and abdominal aortic regions. Single-cell transcriptome analysis revealed that aortic challenge in wild-type mice induced the DNA damage response, the inflammatory response, dedifferentiation and cell death in SMCs, and matrix metalloproteinase expression in macrophages.
…Our findings indicate that the presence of cytosolic DNA and subsequent activation of cytosolic DNA sensing adaptor STING signaling represent a key mechanism in aortic degeneration and that targeting STING may prevent sporadic AAD development.
From: “Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture“ (2019)
The importance of lncRNA H19 and atherosclerosis & aortic dissection
I did a deep dive into SARS-CoV-2 related long non-coding RNA’s in February ‘23:
What are long non-coding RNA’s (lncRNA)?
LncRNAs are a type of RNA generally defined as transcripts of more than 200 nucleotides that are classically considered to not be translated into proteins1, although there is evidence that some peptides may be expressed, but these are considered highly unstable and without biological function2.
I will focus on some of the findings of relevance here, as I can add to them somewhat. Its an area that no regulator will touch - they either don’t know, don’t want to know or are hoping that nobody notices.
Nothing has changed in a year and pharma certainly aren’t going to come clean, we are all just seeing the “unexplained” illnesses and deaths in consequence.
So how is Spike upregulating the MMP’s? The short answer is that Spike mRNA binds to and massively upregulates the lncRNA called H19. This then acts as sponge for microRNA miR-193b-3p. One result of this is that it can no longer restrict gene expression of MMP-2 & MMP-9, which leads to the breakdown of the endothelial wall. Loss of integrity leads to either progression of atherosclerosis or/and arterial wall weakness and aneurysm.
Its not the whole story but an important contributory factor to it.
I prefer to work backwards from pathology to upstream signalling as it is easier to understand in this order rather than starting with a complex, but abstract concept.
Key takes from “Long non-coding RNA H19 in atherosclerosis: what role?” by Shi et al.41 (2020, emphasis mine):
Recent studies suggest that lncRNA-H19 plays important roles in the regulation of angiogenesis, adipocyte differentiation, lipid metabolism, inflammatory response, cellular proliferation and apoptosis.
Some previous trials found that lncRNA-H19 is highly expressed in human atherosclerotic plaques and injured carotid arteries in rat model but was barely expressed in normal coronary arteries (Han et al. 1996; Kim et al. 1994).
VSMC’s: Vascular smooth muscle cells.
Recent studies have shown that increased plasma level of lncRNA-H19 is associated with an increased risk of coronary artery disease (Bitarafan et al. 2019; Zhang et al. 2017). In patients with atherosclerosis, a high level of lncRNA-H19 is detected and overexpression of lncRNA-H19 promotes proliferation and inhibits apoptosis of VSMCs (Pan 2017).
Furthermore, Huang et al. reported that overexpression of lncRNA-H19 contributes to the occurrence of atherosclerosis (Huang et al. 2019). These findings indicate that lncRNA-H19 may be involved in the onset and progression of atherosclerosis.
Green arrows denote promotion, red bars for inhibition:
From: “Fig.1. The role of lncRNA-H19 and its targets in atherosclerosis-related metabolisms. LncRNA-H19 may regulate several targets implicated in the dysregulation of angiogenesis, adipogenesis, lipid accumulation, inflammatory responses, cellular proliferation and apoptosis.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7374855/
Key takes from “LncRNA H19 regulates smooth muscle cell functions and participates in the development of aortic dissection through sponging miR-193b-3p“ by Ren et al.42 (2021 , emphasis mine):
Results:LncRNA H19 was abnormally high-expressed in thoracic aorta tissues of AD patients, and it could competitively bind to and inhibit miR-193b-3p.
HASMC’s: human aortic smooth muscle cells.
PDGF-BB: platelet-derived growth factor BB.
In the PDGF-BB group, the expressions of H19, matrix metallopeptidase (MMP) 2 (MMP-2) and MMP-9 were up-regulated and the expressions of miR-193b-3p, α-SMA, and SM22α were down-regulated; moreover, the proliferation and migration rate of HASMCs were increased.
However, H19 silencing reversed the regulation of PDGF-BB on HASMCs. More interestingly, miR-193b-3p inhibitor could partially reverse the effect of H19 silencing. In addition, the above results were verified by animal experiments, showing that shH19 and up-regulated miR-193b-3p could significantly reduce the thoracic aorta pathological damage in AD mice.
Conclusion: LncRNA H19 regulated smooth muscle cell function by sponging miR-193b-3p and it participated in the development of AD.
Thoracic AD (TAD) is a type of AD, according to pathological morphology [2]. Recent studies found that TAD is a comprehensive pathological change process caused by pathological changes involving multiple blood vessel constituents, such as human aortic smooth muscle cells (HASMCs) and extracellular matrix (ECM) [3,4].
In the pathogenesis of TAD, vascular smooth muscle cells (VSMCs) play an important role in the process of aortic wall contraction and synthesis in the presence of the stimulation of various cells to promote vascular remodeling [5]. With the phenotypic change, VSMCs transform from contraction (differentiation) phenotype to synthesis (dedifferentiation) phenotype [6,7].
Although a large number of studies demonstrated that some factors such as matrix metalloproteinases (MMPs) directly participate in the degradation of the ECM of aorta [9], but the interaction of these factors and their upstream regulatory factors are still not clear.
H19 has tissue specific effects on cancer, either stimulatory or inhibitory. As we see more case reports this is probably one of the contributory factors:
Study found that lncRNA H19 (H19) may play a mediating role between c-Myc and downstream gene expressions in colon cancer [13], and is up-regulated in liver cancer [14], bladder cancer [15] and breast cancer [16], suggesting that H19 may be related to the occurrence of cancer.
From: “Figure 1. LncRNA H19 was abnormally high-expressed in thoracic aorta tissues of AD patients, and could competitively bind and inhibit miR-193b-3p”. Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7823186/
As shown in Figure 4A,B, compared with the Sham group, the expression of H19 in the AD group was significantly up-regulated, but that of miR-193b-3p was greatly down-regulated.
Moreover, injection of AD mice with lentivirus carrying shH19 noticeably up-regulate the miR-193b-3p level (P<0.001). HE staining results showed obvious vascular media degeneration in the AD group, whose muscle fiber assembly was disordered, and the middle membrane thickness in the thoracic aorta increased remarkably (Figure 4C).
However, shH19 treatment can significantly reduce vascular medial degeneration and muscle fiber assembly disorder, and decrease the middle membrane thickness in the thoracic aorta (Figure 4C).
Other regulatory pathways are involved and H19 regulates several processes.
In view of the complexity of the molecular regulatory network in diseases, this may not be the only regulatory pathway of H19. Our research associated H19 with miR-193b-3p for the first time.
The current results showed that H19 competitively bound to and inhibited miR-193b-3p during AD development, suggesting that H19 acts as an adsorbent sponge for miR-193b-3p. Mechanistically, H19 can capture the target sequence of miR-193b-3p and isolate miR-193b-3p from its target mRNA, thereby regulating the function of related physiological pathology in AD.
HIF1a: hypoxia-inducible factor 1-alpha.
The above studies indicate that the function of H19 may be different in different diseases. According to the literature, H19-derived miR-675 directly targets PTEN to stimulate VSMC proliferation, similarly, VSMC apoptosis is induced by H19 via HIF1α [37].
Key takes from “Interplay between SARS‐CoV‐2 and human long non‐coding RNAs“ by Moazzam‐Jazi et al.43 (2021, emphasis mine):
In the present study, using publicly available RNA sequencing data of bronchoalveolar lavage fluid (BALF) and peripheral blood mononuclear cells (PBMC) samples from COVID‐19 patients and healthy individuals, three interesting findings highlighted:
(a) More than half of the interactions between lncRNAs‐PCGs of BALF samples established by three trans‐acting lncRNAs (HOTAIRM1, PVT1 and AL392172.1), which also exhibited the high affinity for binding to the SARS‐CoV‐2 genome, suggesting the major regulatory role of these lncRNAs during the SARS‐CoV‐2 infection.
I have to disagree with this generic statement, especially as their cited study contradicts it:
(b) lncRNAs of MALAT1 and NEAT1 are possibly contributed to the inflammation development in the SARS‐CoV‐2 infected cells. (c) In contrast to the 3′ part of the SARS‐CoV‐2 genome, the 5′ part can interact with many human lncRNAs. Therefore, the mRNA‐based vaccines will not show any side effects because of the off‐label interactions with the human lncRNAs.
In other words lncRNA interactions were never considered and investigated, at least according to published information. So how can they say there will not be side effects?
Overall, the putative functionalities of lncRNAs can be promising to design the non‐coding RNA‐based drugs and to inspect the efficiency of vaccines to overcome the current pandemic.
Again, this uncited statement is flatly contradicted by another study, and uses indefinite terminology:
Also, the viral portion of SARS‐CoV‐2 harbouring the sequence coding spike protein tends to interact neither with human proteins nor with human lncRNAs, implying that the mRNA‐based vaccines will not show the possible side effects because of the off‐label interactions with these macromolecules.
H19 is not referred to in their discussion, which is curious as their supplementary material shows that it is significantly increased in the BALF of COVID patients:
The following year these authors took the time to read the source data. We also know that the synthetic mRNA gene agents also, coincidentally, upregulate these cytokines too4445.
If it’s not due to H19 upregulation then what pathways are being invoked by “vaccination”?
Interestingly, H19 was also found dramatically upregulated in BALF of COVID-19 patients (Moazzam-Jazi et al., 2021). Since H19 is well-known for its role in inflammatory responses [reviewed in (Shi et al., 2020)], the sharp increase in H19 is more likely related to its function of stimulating IL-1β, IL-6 and IL-17 production (Hu et al., 2019; Zhang et al., 2020), and ORF8 hijacking might just be one of the many contributors to the outcome.
From “Assessing the suitability of long non-coding RNAs as therapeutic targets and biomarkers in SARS-CoV-2 infection“ (2022)
Key takes from the somewhat alarming “MicroRNAs and Long Non-Coding RNAs as Potential Candidates to Target Specific Motifs of SARS-CoV-2” by Natarelli et al.46 (2021, a de facto rebuttal of the previous review, emphasis mine):
Here, we report a computational study demonstrating the existence of target motifs in the SARS-CoV-2 genome suitable for specific binding with endogenous human micro and long non-coding RNAs (miRNAs and lncRNAs, respectively), which can, therefore, be considered a conceptual background for the development of miRNA-based drugs against COVID-19.
The SARS-CoV-2 genome contains three motifs in the 5′UTR leader sequence recognized by selective nucleotides within the seed sequence of specific human miRNAs.
The “kill shot”, if that’s the right expression:
Transcript: a length of RNA or DNA that has been transcribed respectively from a DNA or RNA template.
Similarly, lncRNA H19 binds to the 5′UTR of the viral genome and, more specifically, to the transcript of the viral gene Spike, which has a pivotal role in viral infection.
Most lncRNA’s interact weakly, but unfortunately H19 isn’t one of them. It stands out like a beacon of doom for cardiovascular disease and aortic dissection. If it doesn’t interact with synthetic Spike mRNA then someone needs to produce the research, not just arm wave and opine.
They also need to explain case reports.
According to their nuclear localization, we assumed that interaction with SARS-CoV-2 transcripts was unlikely. According to the same computational approach adopted for miRNA BS identification, and considering lncRNA complex secondary structures, lncRNA H19 showed the highest and most significant interaction propensity (IE) with the SARS-CoV-2 5′UTR (−20.82) and Spike mRNA (−40.43) (Figure 5b).
From: “Figure 5. Interaction propensity of lncRNAs involved in pulmonary arterial hypertension, antiviral response, and inflammatory diseases. (a) Heat map of lncRNA interaction propensity with SARS-CoV-2 5′UTR, 3′UTR, and with Spike mRNA using IntaRNA, RNAup, and RNAplex. Significantly binding sites for Spike were identified for cytoplasmic (b) lncRNAs H19 (and for 5′UTR), (c) FENDRR, and (d) LINC01505. Nuclear lncRNAs were used as negative control. Rectangles represent the zoomed view of lncRNA interaction loops. LncRNA minimum free energy secondary structures were predicted using RNAfold web tool. Colors represent base pair probabilities. * p < 0.05; ** p < 0.01; *** p < 0.001.” Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7931055/
Atherosclerosis, SARS-CoV-2 and TLR signalling of MyD88 - a real life Grimm’s fairy tale
I will now hand over the keys to the kingdom to our guest writer, Le Biochimiste, (@c0v1d1984) who did a wonderful job. Enjoy!
Nb. Not for small children’s eyes…
Once Upon a Time…in times of harmony, SIRT1 also diligently performed a duty of deacetylating NF-κB, inhibiting its activity and preventing it from marching into the nucleus and triggering the expression of pro-inflammatory genes. The cellular environment remained peaceful, and the risk of inflammation was kept in check.
However, as SIRT1 depended on NAD+ to carry out its deacetylase activity, SIRT1 struggled to deacetylate NF-κB effectively, making them more transcriptionally active, unleashing a storm of pro-inflammatory genes upon the cellular landscape.
Enter the pro-inflammatory macrophages, the foot soldiers responding to the call of NF-κB. Laden with lipids, these macrophages contributed to the formation of fatty streaks and laid the foundation for early atherosclerotic lesions. The once-harmonious cellular environment transformed into a battleground. The inflammatory signals from activated macrophages attracted other immune cells, fostering an environment ripe for trouble.
Smooth muscle cells, enticed by the inflammatory melody, began to migrate and proliferate, constructing the first bricks of atherosclerotic plaques. Now, a vicious circle took hold: Pro-inflammatory macrophages, entrenched in their mission, perpetuated the inflammatory environment within the arterial walls, and inflammatory signals, in turn, further diminished the already scarce NAD+ levels.
So, brick by brick, atherosclerotic plaques were built, and soon chest pain resonated, and heart attacks unfolded in the realm of the coronary arteries, while the carotid bore witness to transient ischemic attacks and strokes.
The MYD88 gene provides instructions for making a protein involved in signaling within immune cells. The MyD88 protein acts as an adapter, connecting proteins that receive signals from outside the cell to the proteins that relay signals inside the cell. In particular, MyD88 transfers signals from certain proteins called Toll-like receptors and interleukin-1 (IL-1) receptors, which are important for an early immune response to foreign invaders such as bacteria.
In response to signals from these receptors, the MyD88 adapter protein stimulates signaling molecules that turn on a group of interacting proteins known as nuclearfactor-kappa-B. Nuclear factor-kappa-B regulates the activity of multiple genes, including genes that control the body's immune responses and inflammatory reactions. It also protects cells from certain signals that would otherwise cause them to self-destruct (undergo apoptosis).
From “MYD88 gene. MYD88 innate immune signal transduction adaptor”
Yao et al. (2023) discussed how Spike induces overexpression of MyD88. MyD88 acts as a bridge between toll-like receptors (TLRs) and downstream signalling by organising a “myddosome” signalling complex. If this happens in the medial prefrontal cortex (mPFC) of the brain then neuroinflammation and stress susceptibility can be induced, leading to post-COVID (or vax) neuropsychiatric symptoms such as depressive-like behaviour.47
Sahanic et al. (2023) investigated how SARS-CoV-2 induces hyperinflammation via the TLR4/MyD88 pathway and stimulation of macrophages. They found that the virus leads to TLR4 dependent IL-6 upregulation, regardless of variants studied, and the ectodomain (the domain of a membrane protein that extends into the extracellular space) of Spike leads to TLR4 dependent upregulation of IL-6 via the MyD88 pathway.48
In 2022, Chu et al. used animal models of atherosclerosis and foam cell models to demonstrate that miR-146a, IRAK1 and TRAF6 were abnormally expressed in plaques of atherosclerotic animals.
Its not just TLR4 that acts as a receptor. In 2021 Zheng et al. showed that expression of both TLR2 and MYD88 were associated with COVID infection severity. TLR2 signalling via MYD88 is also induced by Spike.49
MYD88 isn’t there only to cause inflammation. In 2008 Sheahan et al. used a mouse model to demonstrate that inhibition of MYD88 leads to 90-100% mortality of SARS-CoV-1 infected mice:
“MyD88−/− mice had significantly higher viral loads in lung tissue throughout the course of infection. Despite increased viral loads, the expression of multiple proinflammatory cytokines and chemokines within lung tissue and recruitment of inflammatory monocytes/macrophages to the lung was severely impaired in MyD88−/− mice compared to wild-type mice.”50
Although a necessary part of anti-Spike or anti-viral immune signalling the relevance to this Substack is that MyD88 contributes to atherosclerosis by recruiting macrophages to the artery wall, using chemokines. Both LPS from microbial pathogens (or due to vax adulteration) and hyperlipidaemia can activate macrophage innate immunity signalling pathways.51
Downstream of TLR4 and MyD88, Chu et al. (2022) discussed how miR-146 is a negative feedback mediator of inflammatory response, which acts as a regulator of atherosclerosis. MiR-146 disrupts the RIG-I pathway by targeting TRAF6, IRAK1, and IRAK2, and negatively regulates RIG-I-dependent type I IFN in macrophages. Most crucially, abnormally higher expression of miR-146 occurs in atherosclerotic plaques than that of non-atherosclerotic arteries.52
Returning to abdominal aortic aneurysms (AAAs) induction mechanisms, OwensIII et al. (2011) used a mouse model to demonstrate how MyD88 and TLRs 2 & 4 contribute to angiotensin II (AngII) induced AAAs and atherosclerosis.53
Figure 1 shows that atherosclerotic lesion area and aortic and thoracic artery diameters correlate positively with MyD88 genotypes (+/+), as well as with apolipoprotein E deficient (apoE−/−) and low-density lipoprotein receptor deficient (LDLR−/−) mice. MyD88 alone can signal these 2 vascular pathologies:
From: “Figure 1. Myeloid differentiation factor 88 (MyD88) deficiency attenuated angiotensin II-induced abdominal aortic aneurysms and atherosclerosis in both apolipoprotein E deficient (apoE−/−) and low-density lipoprotein receptor deficient (LDLR−/−) mice. (A) Maximal external width of abdominal aortas was measured in apoE−/− and LDLR−/− mice that were either MyD88+/+ or −/−. Percent atherosclerotic lesion area was determined on aortic arches and thoracic aortas in apoE−/− (B) and LDLR−/− (C) mice that were either MyD88+/+ or −/−. Triangles represent individual mice, circles represent means, and bars represent SEM. * denotes P<0.001 by Mann-Whitney rank sum analysis.” Source: https://www.ahajournals.org/doi/10.1161/atvbaha.111.238642
Back in 2013, Lunberg et al. used a low-density lipoprotein receptor-deficient (Ldlr−/−) mouse model and bone marrow transplants to show that TLR3 is also a pro-atherogenic receptor in haematopoietic immune cells, and that deletion of either TRIF-related adaptor molecule (TRAM) or TIR-domain-containing adaptor-inducing interferon-β (TRIF) in immune cells was sufficient to reduce vessel inflammation and to protect against atherosclerosis. MyD88 is also crucial for transducing signals from receptors for two well-researched stimuli of atherosclerosis: IL-1 and IL-18.54
S1 Spike alone is sufficient to induce expression of cardiopulmonary-derived IL-18, providing yet another viral/synthetic Spike pathway to accelerated atherosclerosis.55
TL;DR: Due to conflicts of interest the arbitrary thresholds for a diagnosis of hypertension have been coming down at various review meetings over the last 50 years until around 50% of US adults have been diagnosed and around 88% of these have been prescribed blood pressure lowering medications. Under previous thresholds you were healthy but became suddenly sick at the next checkup.56
I know from experience that once in the club that the plan of action is not based on root cause analysis and addressing that, other than the one factor: the long debunked lipid hypothesis. Pass that “test” and you get to look forwards to a lifetime of statins too. In short its a racket and it brings in almost $2 billion per year.57 Statins are an even bigger money spinner and bring in another $15 billion.58
One of the theory’s behind taking blood pressure meds to reduce mortality is that a reduction in hypertension can reduce the incidence or progression of atherosclerosis and other pathologies. In a sense this is correct today, but mainly due to side effects.
Key takes from “Vascular smooth muscle contraction in hypertension” by Touyz et al.59 (2018, emphasis mine):
Hypertension is a major risk factor for many common chronic diseases, such as heart failure, myocardial infarction, stroke, vascular dementia, and chronic kidney disease.
Pathophysiological mechanisms contributing to the development of hypertension include increased vascular resistance, determined in large part by reduced vascular diameter due to increased vascular contraction and arterial remodelling.
These processes are regulated by complex-interacting systems such as the renin-angiotensin-aldosterone system, sympathetic nervous system, immune activation, and oxidative stress, which influence vascular smooth muscle function.
Vascular smooth muscle cells are highly plastic and in pathological conditions undergo phenotypic changes from a contractile to a proliferative state.
Vascular smooth muscle contraction is triggered by an increase in intracellular free calcium concentration ([Ca2+]i), promoting actin–myosin cross-bridge formation.
Growing evidence indicates that contraction is also regulated by calcium-independent mechanisms involving RhoA-Rho kinase, protein Kinase C and mitogen-activated protein kinase signalling, reactive oxygen species, and reorganization of the actin cytoskeleton.
Activation of immune/inflammatory pathways and non-coding RNAs are also emerging as important regulators of vascular function. Vascular smooth muscle cell [Ca2+]i not only determines the contractile state but also influences activity of many calcium-dependent transcription factors and proteins thereby impacting the cellular phenotype and function.
The underpinnings for drugging on such a huge scale are the J-curves. One of the problems of bringing the systolic pressure down to target is that the diastolic may be more responsive and mortality actually increases, and the same is true for systolic below a threshold due to risk of falls and heart failure.
Systolic(SBP) (the first, higher figure as in 120/80 mmHg): A measure of the pressure in your arteries when your heart beats.
Diastolic (DBP): The pressure in your arteries when your heart rests between beats.
Key takes from “The J-curve between blood pressure and coronary artery disease or essential hypertension: exactly how essential?” by Messerli & Panjrath60 (2009, emphasis mine):
The topic of the J-curve relationship between blood pressure and coronary artery disease (CAD) has been the subject of much controversy for the past decades. An inverse relationship between diastolic pressure and adverse cardiac ischemic events (i.e., the lower the diastolic pressure the greater the risk of coronary heart disease and adverse outcomes) has been observed in numerous studies.
This effect is even more pronounced in patients with underlying CAD. Indeed, a J-shaped relationship between diastolic pressure and coronary events was documented in treated patients with CAD in most large trials that scrutinized this relationship.
In contrast to any other vascular bed, the coronary circulation receives its perfusion mostly during diastole; hence, an excessive decrease in diastolic pressure can significantly hamper perfusion.
This adverse effect of too low a diastolic pressure on coronary heart disease leaves the practicing physician with the disturbing possibility that, in patients at risk, lowering blood pressure to levels that prevent stroke or renal disease might actually precipitate myocardial ischemia.
However, these concerns should not deter physicians from pursuing a more aggressive control of hypertension, because currently blood pressure is brought to recommended target levels in only approximately one-third of patients.
Meds may selectively kill only those thought to need them:
In the HOT (Hypertension Optimal Treatment) study, Hansson et al. (20) and Cruickshank (21) scrutinized the high-risk patient group with coronary ischemia. He found that there was a 22% increase in the risk of MI when the DBP was <80 mm Hg compared with <85 mm Hg. The relationship between MI and DBP was J-shaped in patients with coronary ischemia but not in nonischemic patients.
The flatter the systolic curve the less benefit from being medicated, conversely the steeper the diastolic J-curve the greater the risk due to overdosing. The risk profile is asymmetric:
Current guidance is dangerously low for the elderly, according to these curves:
So how did we get here? A key study was the Framingham Heart Study, which is ongoing today:
In 1957, the Framingham Heart Study (N=5,209) reported a threefold higher incidence of atherosclerotic heart disease in 98 adults with a baseline SBP/DBP ≥160/95 mm Hg compared to 310 with a baseline SBP/DBP <140/90 mm Hg16.
From: “Evolution of blood pressure clinical practice guidelines: a personal perspective.“ (2019)
So does targeting symptomatic BP in isolation actually reduce mortality? As we have seen with COVID-19, if you have untreated underlying comorbidities that contribute to this then no, it doesn’t. In fact mortality was greatest in the treated group of a long term statistically powered study, despite their BP being controlled.
Key takes from a Swedish funded study “Survival in treated hypertension: follow up study after two decades” by Andersson et al.61 (1998, emphasis mine):
Objective: To compare survival and cause specific mortality in hypertensive men with non-hypertensive men derived from the same random population, and to study mortality and morbidity from cardiovascular diseases in the hypertensive men in relation to effects on cardiovascular risk factors during 22-23 years of follow up.
Results:Treated hypertensive men had significantly impaired probability of total survival as well as survival from coronary heart disease and stroke.All cause mortality as well as coronary heart disease and stroke mortality were very similar in hypertensive men and normotensive men during the first decade, but increased steadily thereafter despite continuous good blood pressure control.
Apart from risk of stroke, elevated BP wasn’t the cause, it was a symptom. And statins are unlikely to help you here much either, although they have marginal anti-inflammatory effects:6263646566
Smoking, signs of target organ damage, and high serum cholesterol levels, but not blood pressure at screening, were significantly related to the incidence of coronary heart disease during follow up. In time dependent Cox’s regression analysis, the incidence of coronary heart disease was significantly related only to serum cholesterol concentrations in the study.
Conclusion: Treated hypertensive men had impaired survival and increased mortality from cardiovascular disease compared with non-hypertensive men of similar age. These differences were observed during the second decade of follow up. During an observation period of 22-23 years—about 15 000 patient years—hypertensive men receiving diuretics and β blockers had no increased risk of cancer or non-cardiovascular disease.
From: “Figure 1. Cumulative probability of survival of 686 men treated for hypertension and 6810 non-hypertensive men in primary prevention study”. Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC28606/
From: “Figure 2. Cumulative probability of survival from coronary artery disease in 686 men with hypertension and 6810 non-hypertensive men in primary prevention study”. Source: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC28606/
“Beta blockers, also called beta adrenergic blocking agents, block the release of the stress hormones adrenaline and noradrenaline in certain parts of the body. This results in a slowing of the heart rate and reduces the force at which blood is pumped around your body.”67
Beta blockers may, in theory, increase cardiovascular risk due to impaired metabolism of glucose and lipids, but this isn’t the whole story:
Drugs related to metabolism have been previously analysed in the present patient series, but there was no evidence of increased risk of complications from elevated triglyceride concentrations or impaired glucose tolerance induced by treatment.23
Increased rates of mortality seemed to correlate with the administration of the newer generation of drugs:
The treatment in our study may seen outdated, but it should be emphasised that newer treatment with calcium blockers and angiotensin converting enzyme inhibitors became increasingly more common only during the later part of follow up.
It was during this period, however, that the increased morbidity among the hypertensive men was most evident. To date no study has shown the effects of more modern treatment on hard end points.
So if the damage is already done are you “pushing on a string”? The paradox here is that if elevated BP is the trigger for treatment then by the time you are aware of this it may already too late, at least for the types of allopathic meds being prescribed:
It is possible that the preventive effect of lowering blood pressure was less than expected because treatment was started too late, especially as target organ damage (WHO stage 2-3 at the start of intervention) strongly increased the risk of ischaemic heart disease.
Conclusion
Our study of a representative group of hypertensive men showed that despite treatment they had a significantly higher long term mortality from cardiovascular diseases than normotensive men.Part of the risk is irreduciblewith the treatment regimens available at the time of the study,and may be related to cardiovascular damage and risk factors present before established hypertension was diagnosed. During treatment only an elevated serum cholesterol concentration was predictive of coronary heart disease, and high blood pressure levels were associated with an increased risk of stroke.
Glagov’s phenomenon - a challenge for allopathic meds
Glagov’s is an important phenomenon in relation to vascular remodelling in response to the growth of atherosclerotic plaques. I won’t go into a detailed description of all the physics and fluid dynamics, but it’s important to understand the effects.
You quickly understand what a challenge it is to treat hypertension safely once too much damage has occurred and why common meds fail in reality, once away from the case controlled clinical trials.
Due to evolution human beings are surprisingly tolerant of abuse and coping with our unintentional attempts at killing ourselves. Our arteries can cope quite well with atherosclerosis, up to a point. After that the pathophys equivalent of bankruptcy occurs, usually by late middle age.
Glagov’s Phenomenon: A Characteristic Feature of Arterial Remodeling
Atherosclerosis
Although conventional wisdom, before the 1980s, held that atherosclerosis narrows vessels by encroachment of the growing plaque into the lumen, Glagov and colleagues1 found that the lumen area of atherosclerotic human coronaries remained constant until the percent stenosis exceeded 40% (Figure 1B).
At this point lumen diameter decreased, resulting in a restriction in flow (Figure 1B, right). Glagov’s postmortem study in humans was consistent with prior data obtained in animals with experimentally induced atherosclerosis.9,10
In particular, Bond and colleagues9 noted that cynomolgus monkey fed an atherosclerotic diet for 3 years exhibited not only greater lesions, but also much larger coronary internal elastic lamina (IEL) compared with animals on control diet.
Armstrong et al10 further showed that iliac-femoral arteries enlarge with preserved lumen in atherosclerotic rhesus and cynomolgus monkeys. More recently, Glagov’s phenomenon has been observed in atherosclerotic mice.11
From: “Figure 1. Scheme for vascular remodeling. A, Normal artery. White arrow points to physiological remodeling, black arrow to pathophysiological remodeling. B, Progression of atherosclerosis causes lumen narrowing when stenosis exceeds 40% (adapted from1). C, Vascular injury after percutaneous transluminal angioplasty (PTCA) causes constrictive remodeling with decreased vessel size (restenosis), whereas probucol treatment promoted outward vessel remodeling and prevented lumen narrowing (No restenosis: −0.2 mm in probucol versus −1.2 mm in placebo, a 6-fold inhibition of restenosis). However, vessel wall increased equally (0.3 mm) in both groups.14,15” Source: https://www.ahajournals.org/doi/10.1161/ATVBAHA.106.129254
Why lipid lowering and anti-hypertensive drug trials fail in the real world is that, although meeting their goals for efficacy in plaque reduction this doesn’t necessarily translate into Glagovian atheroregression.
In 2015 Alexander N Kharlamov of the De Haar Research Foundation wrote an excellent special report about the challenge. Its easier to demonstrate regression from your novel drug in small, early stage lesions that aren’t a risk to health than late stage necrotic and calcified fibroatheromas, which certainly are.
The modern-day statin trials were conducted in patients with relatively small lesions when PAV never exceeded 40% and without proper methodology, which means that the revealed phenomenon of atheroregression is essentially the pseudo reduction of the atheroma volume.
The PAV or plaque burden is the only parameter to describe changes in the vessel geometry from the Glagov phenomenon point of view because it mathematically reflects patterns between both vessel and lumen size.
…Prevention of atherosclerosis and treatment of its complications remain a clinical challenge [1]. Some recent clinical trials demonstrated [1–10] moderate atheroprotective effect of the different lipid-lowering medications. There are some achievements as well as methodological flaws, which require our attention in order to optimize the research tools for imaging and treatment in interventional cardiology with the final goal to reverse Glagov atherogenesis.
…Early stages of lesion development may be associated with overcompensation. At more than 40% stenosis, however, the plaque area continues to increase to involve the entire circumference of the vessel, and the artery no longer enlarges at a rate sufficient to prevent narrowing of the lumen.
…At present, although atheroregression below the Glagovian threshold has not been yet achieved, developments in nanotechnologies may ultimately realize this goal.
…In most statin trials (Figure 4), the baseline PB was below 40% which means already beyond Glagov threshold. These lesions cannot be interpreted as fibroatheromas, and, moreover, PROSPECT study [24] affirmed a 70% PB as the independent predictor of the major cardiac adverse events in non-culprit lesions, which means we cannot expect any major outcomes in those trials either. In that case here is a question what we try to achieve in such young and most probably clinically silent lesions.
On the one hand, plaques at those trials could be characterized as relatively small and early-stage lesions that a priori makes them more sensitive to the intensive drug therapy in comparison with the late-stage and advanced fibroatheromas with pronounced inorganic component.
On the other hand, these tiny lesions depreciate the atheroprotective potential of the drug agent or device due to low initial vessel volume, which means the clinical value of the approach could be merely underestimated.
From: “Why do we fail to achieve Glagovian atheroregression in lipid-lowering trials?“ (2015)
“ACE inhibitors can reduce the activity of an enzyme called angiotensin-converting enzyme, or ACE for short. The enzyme is responsible for hormones that help control your blood pressure. It has a powerful narrowing effect on your blood vessels, which increases your blood pressure. ACE inhibitors inhibit or limit this enzyme, making your blood vessels relax and widen. This, in turn, lowers your blood pressure and improves blood flow to your heart muscle.”68
Briefly referred to above, how effective are these at reducing mortality rates? As antihypertensive drugs they are ironically quite ineffective at sustainably reducing BP by a significant amount and the side effects profiles are notable too. But mortality rates do appear to be significantly lower. Of course with fraud being so rampant in pharma funded trials we do need to take all such mortality data with a pinch of lo-salt.
OBP is a great example of inaccuracies feeding into sales. How many prescriptions are made based on these measurements, and these alone?
Office blood pressure (OBP): “For almost a century, office blood pressure (OBP) measurement has been regarded as the cornerstone for diagnosis and management of hypertension because the vast majority of the evidence on the risks associated with elevated BP and the benefits of treatment-induced BP lowering has been based on OBP measurements…OBP is confounded by the white coat and the masked hypertension phenomena, inaccurate devices, and observer-related factors, such as imperfect methodology, observer error and bias, and failure to standardize the circumstances of measurement….At the present time, and for some time to come, it is likely that the diagnosis and management of hypertension in most people will be based on OBP measurement. However, even the most standardized OBP can be misleading in a considerable proportion of subjects, and out-of-office BP measurements should be used before making a diagnosis of hypertension and to guide treatment.1 In case of disagreement between OBP and out-of-office BP measurements (ambulatory or home), decisions should always be based on the latter.”69
Whether OBP provides an accurate diagnosis or not either way ramiprilappears to have a negligible effect on OBP, according to another Swedish study: “Comparative Effects of Ramipril on Ambulatory and Office Blood Pressures, A HOPE Substudy” by Svensson et al.70 (2001, emphasis mine):
In the HOPE-trial, the ACE inhibitor ramipril significantly reduced cardiovascular morbidity and mortality in patients at high risk for cardiovascular events. The benefit could only partly be attributed to the modest mean reduction of office blood pressure (OBP) during the study period (3/2 mm Hg).
However, because according to the HOPE protocol ramipril was given once daily at bedtime and blood pressure was measured during the day, the 24-hour reduction of blood pressure may be underestimated based on OBP. Thirty-eight patients with peripheral arterial disease enrolled in the HOPE study underwent 24-hour ambulatory blood pressure (ABP) measurement before randomization and after 1 year.
BP was mainly reduced at night, but with little effect on OBP as the effects peak 3-6 hours after taking.
Nb. 139/79mm is classed as high according to current thresholds:
OBP was measured in the sitting position immediately before fitting the ABP measuring equipment to the patients. Ramipril did not significantly reduce OBP (8/2 mm Hg, P=NS) or day ABP (6/2 mm Hg, P=NS) after 1 year.Twenty-four–hour ABP was significantly reduced (10/4 mm Hg, P=0.03), mainly because of a more pronounced blood pressure lowering effect during nighttime (17/8 mm Hg, P<0.001).
Because the mean reduction of office blood pressure (OBP) with treatment was small (3/2 mm Hg) and the majority of patients already had well-controlled blood pressure at baseline (139/79 mm Hg),1 the benefit has been attributed mainly to other mechanisms of ACE inhibition than to blood pressure reduction per se. Such mechanisms could include beneficial effects on the endothelium or anti-atherosclerotic effects.2
From: “Figure 1. Hourly means of systolic and diastolic ABP in the ramipril group (A, n=20) and placebo group (B, n=18) at baseline and 1 year. X-axis indicates time in hours starting at 0:00 am.” Source: https://www.ahajournals.org/doi/full/10.1161/hy1101.099502
Many patients may take their daily dose at bedtime to minimise the impact of side effects such as tiredness, but by the morning the effects have largely worn off:
The pronounced blood pressure–lowering effect during nighttime may be explained by the fact that ramipril was given at bedtime. Ramipril has been reported to have a rather smooth effect on 24-hour blood pressure and can therefore be taken once daily for treatment of hypertension.6, As with all antihypertensive agents, however, there is a variation of the blood pressure–lowering effect during 24 hours, with a peak effect of ramipril 3 to 6 hours after administration.8,9
The mean reduction of OBP that was achieved in HOPE overall throughout the study period (≈3/2 mm Hg) would, compared with previous hypertension trials,10,11 explain a reduction of stroke incidence of ≈13% and a reduction of myocardial infarctions of ≈5%. In HOPE, however, the relative reductions of these endpoints were 31% and 20%, respectively. Therefore, it has been claimed that other vascular protective effects of ACE inhibition, besides blood pressure reduction per se, most probably contributed to the positive effects.
…the findings of this study suggest that the mode of blood pressure measurement and the timing of drug intake in relation to blood pressure measurement are important factors to consider when assessing blood pressure effects and relating these to clinical outcomes.
A Cochrane review from 2008 conducted a meta-analysis (emphasis mine)71:
The class of drugs called ACE inhibitors is commonly used for the treatment of elevated blood pressure. This class includes drugs such as ramipril (brand name: Altace), captopril (Capoten), enalapril (Vasotec), fosinopril (Monopril), lisinopril (Prinivil, Zestril) and quinapril (Accupril).
We found 92 trials that randomly assigned participants to take either an ACE inhibitor or an inert substance (placebo). These trials evaluated the blood pressure lowering ability of 14 different ACE inhibitors in 12 954 participants. The trials followed participants for approximately 6 weeks (though people are typically expected to take anti-hypertension drugs for the rest of their lives).
The blood pressure lowering effect was modest.There was an 8-point reduction in the upper number that signifies the systolic pressure and a 5-point reduction in the lower number that signifies the diastolic pressure.
Most of the blood pressure lowering effect (about 70%) can be achieved with the lowest recommended dose of the drugs. No ACE inhibitor drug appears to be any better or worse than others in terms of blood pressure lowering ability.
Most of the trials in this review were funded by companies that make ACE inhibitors and serious adverse effects were not reported by the authors of many of these trials. This could mean that the drug companies are withholding unfavorable findings related to their drugs.
Due to incomplete reporting of the number of participants who dropped out of the trials due to adverse drug reactions, as well as the short duration of these trials, this review could not provide a good estimate of the harms associated with this class of drugs.
Prescribing the least expensive ACE inhibitor in lower doses will lead to substantial cost savings, and possibly a reduction in dose-related adverse events.
Most MD’s will start at the lowest dose and increase it from there until the target reduction in BP is achieved, or try a stronger drug. According to Cochrane this may be the wrong approach and just serve to disproportionately increase side effects.
Common side effects of ramipril, affecting more than 1% of users include:7273
Weakness or tiredness.
Dizziness.
Chest pain.
Nausea.
Diarrhoea.
A dry, tickly cough that does not get better.
Headaches.
A mild skin rash.
Blurred vision.
Rarer side effects, warranting immediate medical attention:
The whites of your eyes turn yellow, or your skin turns yellow although this may be less obvious on brown or black skin – this can be a sign of liver problems.
You're paler than usual, you feel tired, faint or dizzy, you have any sign of bleeding (like bleeding from the gums and bruising more easily than usual), a sore throat, a high temperature, or you get infections more easily – these can be signs of a blood or bone marrow disorder.
You have severe stomach pain – this can be a sign of an inflamed pancreas (acute pancreatitis).
You have swollen ankles or blood in your pee or you're not peeing at all – these can be signs of kidney problems.
A better way
Sure the NHS (National Help-pharma Service) and similar bodies do not have the time, resources or drive to practice whole health medicine but they could do more to improve outcomes by looking beyond just the one metric determinant for medication. And that’s without even touching on their complicity in the LNP/synthetic mRNA scandal.
Kannel was signposting where to look and where to treat almost a quarter century ago.
Key takes from “Risk stratification in hypertension: new insights from the Framingham Study”74, (2020, emphasis mine):
Although clinicians favor the diagnosis and treatment of hypertension in terms of diastolic blood pressure elevation and categoric cut points, epidemiologic data show a more important influence of systolic blood pressure, and a continuous, graded influence of blood pressure even within what is regarded as the normotensive range.
An important revelation in epidemiologic hypertension research is that hypertension usually occurs in conjunction with other metabolically linked risk factors; therefore, less than 20% occurs in isolation.
The other risk factors that tend to accompany hypertension include glucose intolerance, obesity, left ventricular hypertrophy, and dislipidemia (elevated total, LDL, and small dense LDL cholesterol levels, raised triglyceride, and reduced HDL cholesterol levels).
Clusters of three or more of these additional risk factors occur at four times the rate expected by chance.This clustering is attributed to an insulin resistance syndrome promoted by abdominal obesity. The amount of risk factor clustering accompanying elevated blood pressure was observed to increase with weight gain.
Based on Framingham Study data the prevalence of insulin resistance syndrome in the general population could be as high as 22% in men and 27% in women. Risk of coronary disease, the most common and most lethal sequel to hypertension, increased stepwise with the extent of risk factor clustering.
Among persons with hypertension, about 40% of coronary events in men and 68% in women are attributable to the presence of two or more additional risk factors. Only 14% of coronary events in hypertensive men and 5% of those in hypertensive women occurred in the absence of additional risk factors.
Hypertensive patients are more appropriately targeted for therapy by such risk stratification and the goal of the therapy should be to improve the multivariate risk profile.
Credit where it is due, the NHS is improving in this area.
Please note that they are “very busy at this time”, for some reason:
How to access the programme
The NHS Digital Weight Management Programme could help you if you are living with obesity and also have diabetes, high blood pressure, or both. If this is you, you could benefit from this 12-week programme. It’s free and easily available via a smartphone, tablet, or computer.
How to start the programme
To start your journey to a healthier lifestyle, you need to speak to your GP or a local pharmacist who can refer you to the programme.
Please note that GP and pharmacy teams may be very busy at this time.
Targeting BP as a proxy for diagnosing and treating cardiovascular disease is like the tail wagging the dog.
A disproportionate fall in mortality in response to a small fall in BP is often attributed to the great benefits of BP reducing antihypertensive drugs, but this may be a mirage and more consequential in nature than causative.
Vascular remodelling, as we have just seen, is a consequence of chronic hypertension and not easily reversed just by reducing BP:
Imbalance in matrix metalloproteinase/tissue inhibitors of metalloproteinases may contribute to alteration in collagen turnover and extracellular matrix remodeling. Chronic vasoconstriction may lead to embedding of the contracted vessel structure in a remodeled extracellular matrix, contributing to the inward remodeling of the blood vessel as smooth muscle cells are rearranged around a smaller lumen. The resulting remodeling of small arteries may initially be adaptive, but eventually it becomes maladaptive and compromises organ function, contributing to cardiovascular complications of hypertension.
From: “Vascular Remodeling in Hypertension, Roles of Apoptosis, Inflammation, and Fibrosis“ (2001)
If decreased mortality is not primarily due to significant BP reduction (trial fraud aside), then what is the mode of action of ACE inhibitors? This is thought to be due to pleiotropic anti-inflammatory effects, ie all of a drug's actions other than those for which the agent was specifically developed.
Key takes from “Pleiotropic effects of angiotensin-converting enzyme inhibitors in normotensive patients with coronary artery disease” by Krysiak & Okopień75, (2008, emphasis mine):
The purpose of this study was to compare the effects of plasma- and tissue-type angiotensin-converting enzyme inhibitors on blood pressure and on systemic inflammation, hemostasis and oxidative functions in normotensive patients with stable coronary artery disease.
Plasma lipid profile and the levels of oxidized low density lipoproteins (LDLs), monocyte chemoattractant protein (MCP)-1, interleukin-10, C-reactive protein (CRP), fibrinogen and plasminogen activator inhibitor (PAI)-1 were determined at the beginning of the study and after 30 and 90 days of treatment.
Neither enalapril nor perindopril affected blood pressure or plasma lipids.
Perindopril appeared to have significant cardiovascular benefits, yet BP or lipids weren’t reduced at all.
IL-10 is a potent anti-inflammatory cytokine:
Perindopril significantly reduced plasma levels of oxidized LDLs, CRP, MCP-1, fibrinogen and PAI-1, and increased interleukin-10. The effect of enalapril on these markers of systemic inflammation, hemostasis and oxidative functions was much less pronounced.
The results showed that enalapril and perindopril weredevoid of a blood pressure-lowering effect in normotensive patients with stable coronary artery disease.
Perindopril was superior to enalapril in exhibiting antioxidant, antithrombotic and profibrinolytic activities.
The treatment-induced changes in the balance between pro- and antiinflammatory cytokines and in hemostasis may contribute to the clinical effectiveness of tissue angiotensin-converting enzyme inhibitors in the therapy of atherosclerosis-related disorders.
An interesting review from 2018 by Ahn et al. reported on the effects of enalapril on the 25 year survival of an initial cohort of 558 Belgian patients with left ventricular systolic dysfunction (LVSD).76
Since the study closed 22.1 years earlier 91% of patients had died (398 in prevention arm and 160 in treatment arm). Comparisons to the control group demonstrated a reduction in all-cause mortality from 94% to 88% (p=0.02). A relative risk reduction in death vs placebo of 20% was observed, when data was stratified by trial arms. Long term mortality comparison vs control was 84% vs 92% and post-trial use of ACE inhibitors was similar in both arms at 74%.
These results demonstrated a significant reduction in mortality rates vs control despite enalapril not being associated with significant reductions in either BP or plasma lipids.
7 year comparative data for Ramipril is similar. This study had an initial 9297 patients and 4528 in the follow-up. Analysis revealed a 19% relative risk (RR) reduction of myocardial infarction (95% CI 0.65-1.01); a 16% lower RR of revascularization (95% CI 0.70-0.99) and a 34% lower RR of new onset diabetes (95% CI 0.46-0.95):77
The cure for high blood pressure is high blood pressure
Slightly tongue in cheek. You’ve just been diagnosed, prescribed ACE inhibitors, advised to stop smoking, de-stress, reduce your drinking, improve your diet, lose some weight and…get more exercise!
I fully agree with this but it is somewhat ironic that bursts of intense or sustained moderate exercise to elevate your heart rate (and therefore BP) to a percentage of its maximum for up to 5 hours a week is recommended for maximum health benefits.78 Not all BP elevation is bad BP elevation:
For moderate-intensity physical activity, your target heart rate should be between 64% and 76%1,2 of your maximum heart rate. You can estimate your maximum heart rate based on your age. To estimate your maximum age-related heart rate, subtract your age from 220. For example, for a 50-year-old person, the estimated maximum age-related heart rate would be calculated as 220 – 50 years = 170 beats per minute (bpm). The 64% and 76% levels would be:
64% level: 170 x 0.64 = 109 bpm, and
76% level: 170 x 0.76 = 129 bpm
This shows that moderate-intensity physical activity for a 50-year-old person will require that the heart rate remains between 109 and 129 bpm during physical activity.
For vigorous-intensity physical activity, your target heart rate should be between 77% and 93%1,2 of your maximum heart rate. To figure out this range, follow the same formula used above, except change “64 and 76%” to “77 and 93%”. For example, for a 35-year-old person, the estimated maximum age-related heart rate would be calculated as 220 – 35 years = 185 beats per minute (bpm). The 77% and 93% levels would be:
77% level: 185 x 0.77 = 142 bpm, and
93% level: 185 x 0.93 = 172 bpm
This shows that vigorous-intensity physical activity for a 35-year-old person will require that the heart rate remains between 142 and 172 bpm during physical activity.
From: “Target Heart Rate and Estimated Maximum Heart Rate“
A different approach is to target plaques directly. Hat tip to Moriarty, who recently discussed how in an vitro trial of the drug ABT-263 on atherosclerotic mice the mortality rate went up rather than down, as it reduced indices of plaque stability.
It acts as a senolytic agent, selectively inducing the death of senescent cells which accumulate with age.
ABT-263 treatment also reduced α-SMA+ fibrous cap thickness by 60% and was associated with a > 50% mortality rate. Taken together, ABT-263 treatment of WD-fed Apoe-/- mice with advanced lesions resulted in multiple detrimental changes, including reduced indices of stability and increased mortality
From: “Treatment of advanced atherosclerotic mice with ABT-263 reduced indices of plaque stability and increased mortality“ (2024)
Commercially it is known as Navitoclax and it something of a solution in search of a problem and money has been thrown at it for over 2 decades.
It started out as Bcl-2 inhibiting anti-cancer drug but got discontinued due to its affinity for an essential platelet protein Bcl-XL, leading to thrombocytopenia. Oops.
Reborn for a while its latest flop had been as a treatment for myelofibrosis, a rare blood cancer and they have given up on it now, instead modifying it to a new compound ABT-199. Hopefully it will be associated with less cancer patient deaths.7980
Broad spectrum therapeutics (BSTs)
Most of our BST’s show at least some evidence of anti-atherosclerotic properties. I will focus on some of the more well known meds here, but by all means add more in the comments. I’ve not checked my TCM repository yet but it will have many more.
What they do have in common is that virtually none of them are likely to have been recommended to you, let alone prescribed.
Newer studies should be more up to date, comprehensive and accurate, but unfortunately it doesn’t always work that way, especially after around 2020. There is certainly a place for older studies, especially those involving pure research into signalling pathways and cellular effects.
Another consideration is that absence of evidence of benefits isn’t evidence of absence. There is no reason why correcting deficiency isn’t a highly beneficial treatment goal in itself.
This is a popular dodge used by pharma: if you are not deficient in the trial then adding more doesn’t appear to confer any benefits, therefore it doesn’t work.
Please address via diet as a starting point wherever possible.
Vitamin B6 + B12 + folic acid:
High-dose B-vitamin supplementation significantly reduces progression of early stage subclinical atherosclerosis (carotid artery intima-media thickness) in well-nourished healthy B-vitamin “replete” individuals at low-risk for CVD with a fasting tHcy >9.1 μmol/L.
“High-Dose B-Vitamin Supplementation and Progression of Subclinical Atherosclerosis: A Randomized Controlled Trial” (2008)
The natural antioxidant system includes enzymes and vitamins A, E, and C. The lipophilic vitamins A and E protect the fatty acid components of lipoproteins and membranes, and vitamin C functions in the aqueous phase both directly and by regenerating oxidized vitamin E. In animal models, the antioxidant vitamins protect lipids and prevent atherosclerosis.Population studies suggest an inverse relationship between atherosclerosis and vitamin levels. Several observational studies and some clinical trials have demonstrated that antioxidant vitamin supplements may prevent atherosclerosis.
“The antioxidant vitamins: impact on atherosclerosis” (1993)
The association between vitamin D deficiency and atherosclerosis has been extensively studied. Observational studies consistently report an inverse relationship between vitamin D levels, atherosclerotic risk factors, and markers of subclinical atherosclerosis. Mechanistically, vitamin D exerts anti-inflammatory effects, modulates immune responses, improves endothelial function, and influences lipid metabolism, all of which play critical roles in atherosclerosis development and plaque stability.
“Exploring the Role of Vitamin D in Atherosclerosis and Its Impact on Cardiovascular Events: A Comprehensive Review” (2023)
Prescriptions of this would save far more lives than the current approach.
In an ideal world, Vitamin K2 would have the same association with cardiovascular health that folic acid has with pregnancy. Optimal Vitamin K2 intake is crucial to avoid the calcium plaque buildup of atherosclerosis, thus keeping the risk and rate of calcification as low as possible.1-3
Matrix GLA protein (MGP)—found in the tissues of the heart, kidneys, and lungs—plays a dominant role in vascular calcium metabolism. Its production is stimulated by Vitamin D3, but it requires adequate Vitamin K2 intakes to be activated (similar to the bone-building protein osteocalcin). Once activated by Vitamin K2, MGP can bind calcium and escort it out of the areas where this mineral is destructive, namely arteries and soft tissues.1,4
No other productive mechanism for maintaining flexible blood vessels walls has been discovered, which makes MGP the only known and most potent existing inhibitor of cardiovascular calcification.
That is why Vitamin K2 is crucial as a cardiovascular health nutrient. Here we will endeavor to clear up considerable confusion about Vitamin K2, ensuring the right form is identified, as well as provide the substantial body of evidence confirming its role as a cardiovascular-support nutrient.
“Highlighting The Substantial Body Of Evidence Confirming The Importance Of Vitamin K2 As A Cardio-Support Nutrient, And How The Right K2 Makes All The Difference” (2019)
During the last decade, the combination of Se (Se-enriched yeast 200 µg/day) and coenzyme Q10 (200 mg/day) supplementation [92] to improve cardiovascular health was thoroughly investigated in a long-term study with elderly Swedish citizens. Over 5 years, the reduction of cardiovascular mortality was accompanied by an improvement of N-terminal-pro hormone BNP (NT-proBNP) (a heart failure marker) levels and on echocardiography [93], C-reactive protein and sP-selectin levels [94], myocardial function and reduced fibrosis through the reduction of inflammation and oxidative stress [95].Interestingly, the reduced cardiovascular mortality was observed even at 10 [96] and 12 years [97] follow-up. Positive effects were also observed for the plasma levels of von Willebrand factor (vWf) and the plasminogen activator inhibitor-1 (PAI-1) [98], which are markers of coronary endothelial and ventricular dysfunction and myocardial infarction [99,100].
“The Role of Selenium in Atherosclerosis Development, Progression, Prevention and Treatment” (2023)
Ubiquinone or coenzyme Q10 is a multifunctional molecule that could theoretically revert most of the cellular alterations found in atherosclerosis, such as cholesterol biosynthesis dysregulation, impaired autophagy flux and mitochondrial dysfunction thanks to its redox and signaling properties.
…CoQ levels decline with aging may explain the postulated increase in ROS production with advanced age.
FMD: flow-mediated dilation, the dilation of an artery when blood flow increases in that artery. The primary cause is release of nitric oxide and it helps to avoid hypertension.
CIMT: carotid intima media thickness.
Seven RCTs with 306 participants were included. Mg supplementation significantly increased FMD (MD: 2.97; 95% CI: 0.23 to 5.70%, p = 0.033). Between studies heterogeneity was high and subgroup analysis could not identify the sources of heterogeneity. Magnesium supplementation had no significant effect on CIMT (MD: −0.13 mm; 95% CI: 0.27, 0.01; p = 0.077) with high heterogeneity. Mg dose, duration of treatment, healthy status, baseline CIMT and sample size were the potential sources of heterogeneity. Mg supplementation could decrease CIMT to a greater extent in hemodialysis (HD) patients; lower doses of Mg, higher sample size and follow up duration and subjects with higher baseline CIMT also reduced the heterogeneity to some degree (p < 0.001).
Conclusions
Magnesium supplementation may improve endothelial function without affecting carotid intima media thickness.
“Effect of magnesium supplementation on endothelial function: A systematic review and meta-analysis of randomized controlled trials” (2018)
Purpose: Zinc is considered protective against atherosclerosis; however, the association between dietary zinc intake and cardiovascular disease remains debated. We investigated whether dietary zinc intake was associated with coronary artery calcium (CAC) progression in the Multi-Ethnic Study of Atherosclerosis (MESA).
Results: Dietary zinc intake was 8.4 ± 4.5 mg/day and 2537 (48.9%) of the included participants had CAC at baseline. Over a median follow-up of 3.4 years (25th-75th percentiles = 2.0-9.1 years), 2704 (52.1%) participants had CAC progression. In the fully adjusted model, higher dietary zinc was associated with a lower risk of CAC progression in both men (hazard ratio [HR] 0.697, 95% confidence interval [CI] 0.553-0.878; p = 0.002) and women (HR 0.675; 95% CI 0.496-0.919; p = 0.012, both comparing extreme groups). Furthermore, such an inverse association was attributable to dietary zinc intake from non-red meat (p < 0.05), rather than red meat sources (p > 0.05).
Conclusions: In this multiethnic population free of clinically apparent cardiovascular disease, higher dietary zinc intake from non-red meat sources was independently associated with a lower risk of CAC progression.
“Association of dietary zinc intake with coronary artery calcium progression: the Multi-Ethnic Study of Atherosclerosis (MESA)” (2021)
Methods: Among the 4,266,268 participants without a history of CVD or previous prescription of aspirin and other antiplatelet agents who were screened between 2002 and 2008, 268,963 persons who were prescribed low-dose aspirin (≤100 mg/day) over 90 days in 2002-2008 and 1,075,852 persons who did not receive aspirin were selected after propensity score matching. A Cox proportional-hazards model was used to evaluate the effect of low-dose aspirin on the development of CVD and bleeding episodes.
Results: Aspirin showed a protective effect on total CVD events (hazard ratio (HR); 0.737, 95% confidence interval; 0.729-0.745).The protective effect of aspirin on total CVD events was significant in men, women and even in young participants (<65 years). Aspirin had a protective effect in participants with diabetes or hypertension against all subcategories of CVD. The HR of bleeding risk was 1.4-1.5 in aspirin group.
Conclusions: Low-dose aspirin generally showed a protective effect against CVD regardless of age, sex, and underlying comorbidities in the real world. Though, the effect of aspirin was evident at a young age, the risk of bleeding was also high (1.4-1.5 times), and thus, careful prescription is required.
“Low-dose aspirin in the primary prevention of cardiovascular diseases: A retrospective, propensity score matched study“ (2023)
Recent studying found that berberine could inhibit both the proliferation and apoptosis of VSMCs induced by mechanical stretch stress simultaneously, which suggests that berberine might be an excellent drug to treat atherosclerosis.
…Statins are widely used as the first-line drugs to treat atherosclerosis because of their excellent lipid-lowering functions. However, some patients treated with statins do not achieve the expected efficacy and might have side effects, such as blood glucose changes and hepatotoxicity. For these patients who experience unsatisfactory lipid-lowering effects, new alternatives are urgently needed.
…Berberine [5, 6-dihydro-9, 10-dimethoxybenzo(g)-1, 3-benzodioxolo(5,6-a) quinolizinium, or C20H18NO4], a benzylisoquinoline alkaloid isolated from several Chinese herbal substances, has been widely used in the treatment of inflammatory disorders, microbial and protozoal infections, and intestinal diseases in Ayurvedic medicine and in traditional Chinese medicine for centuries.
…Despite its extremely low plasma concentration, berberine conveys significant pharmacological actions against CVD. Several studies have demonstrated the therapeutic effects of berberine on multiple vessel cells in CVD and will be discussed in Berberine Inhibits EC Dysfunction through Effect of Berberine on VSCs sections.
Garlic indirectly effects atherosclerosis by reduction of hyperlipidemia, hypertension, and probably diabetes mellitus and prevents thrombus formation. In addition, in animal models, garlic causes direct antiatherogenic (preventive) and antiatherosclerotic (causing regression) effects at the level of artery wall. Garlic's direct effect on atherosclerosis may be explained by its capacity to reduce lipid content in arterial cells and to prevent intracellular lipid accumulation. This effect, in turn, is accompanied by other atherosclerotic manifestations, i.e., stimulation of cell proliferation and extracellular matrix synthesis.
We found that treatment with artemisinin (50 and 100 mg/kg) effectively ameliorated atherosclerotic lesions, such as foam cell formation, hyperplasia and fibrosis in the aortic intima. Atherosclerotic mice treated with artemisinin showed reduced inflammation by up-regulating adenosine 5'-monophosphate (AMP) activated protein kinase (AMPK) activation and by down-regulating nuclear factor-κB (NF-κB) phosphorylation and nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome expression in the aortas. In addition, artemisinin was found to promote AMPK activity in macrophages and its anti-inflammatory effect was neutralised by AMPK silence using specific siRNA. In conclusion, we demonstrate that artemisinin may protect the aortas from atherosclerotic lesions by suppression of inflammatory reaction via AMPK/NF-κB/NLRP3 inflammasomes signalling in macrophages.
“Artemisinin alleviates atherosclerotic lesion by reducing macrophage inflammation via regulation of AMPK/NF-κB/NLRP3 inflammasomes pathway” (2020)
Dietary saponins have the potential to ameliorate atherosclerosis (AS). Gypenosides of Gynostemma pentaphyllum (GPs) have been used as functional foods to exhibit antiatherosclerotic activity. The present study aimed to explore the protective effect, underlying mechanism and active substances of GPs on AS in vivo and in vitro. Results demonstrated GPs administration reduced the serum concentrations of TC and LDL-C, upregulated the plasma HDL-C content, inhibited the secretion of ICAM-1, VCAM-1, and MCP-1, and alleviated vascular lesions in VitD3 plus high cholesterol diet-induced AS rats as well as reduced adhesion factors levels in ox-LDL-stimulated HUVECs, which was potentially associated with suppressing PCSK9/LOX-1 pathway.
“Chemical Characterization and Atherosclerosis Alleviation Effects of Gypenosides from Gynostemma pentaphyllum through Ameliorating Endothelial Dysfunction via the PCSK9/LOX-1 Pathway” (2022)
The expression of lipolysis related proteins (PPARα, CPT-1) was increased while the expression of adipogenesis related proteins (SREBP-1c, ACS) was decreased by baicalin treatment, indicating the anti-adipogenic effect of baicalin. Moreover, baicalin up-regulated the activities of antioxidant enzymes (SOD, CAT and GSH-Px) and down-regulated the activity of oxidative parameter MDA compared with AS model group, indicating the anti-oxidant effect of baicalin. The increased levels of pro-inflammatory cytokines (IL-6, TNF-α, sVE-cadherin) induced by AS were also decreased by baicalin treatment, indicating that baicalin acted as an anti-inflammation regulator in AS.
“Baicalin alleviates atherosclerosis by relieving oxidative stress and inflammatory responses via inactivating the NF-κB and p38 MAPK signaling pathways” (2018)
Quercetin (QC) is a natural polyphenolic bioflavonoid with potent antioxidant and anti-inflammatory properties but lacks of clinical attraction due to low oral bioavailability. Piperine is a well established bioavailability enhancer used pre-clinically to improve the bioavailability of antioxidants (e.g., Quercetin).
“Neuroprotective potential of Quercetin in combination with piperine against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity” (2017)
Results: Ox-LDL induced RAW264.7 macrophage-derived foam cell formation, reduced survival, aggravated cell lipid accumulation, and induced a senescence phenotype. This was accompanied by decreased formation of autophagosome; increased expression of P53, P21, and P16; and decreased expression of LC3-II/I and Beclin1. After intervention with quercetin, the cell survival rate was increased, and lipid accumulation and senescence phenotype were reduced. Furthermore, the expression of LC3-II/I and Beclin1 were increased, which was consistent with the ability of quercetin to promote autophagy. Ox-LDL also increased the expression of MST1, and this increase was blocked by quercetin, which provided a potential mechanism by which quercetin may protect foam cells against age-related detrimental effects.
Conclusion: Quercetin can inhibit the formation of foam cells induced by ox-LDL and delay senescence. The mechanism may be related to the regulation of MST1-mediated autophagy of RAW264.7 cells.
“Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells” (2019)
3.3. Protective Effects of Curcumin in Atherosclerosis
The possible atheroprotective properties of curcumin have been studied in many different animal models of atherosclerosis, including Apoe- and Ldlr-deficient mice or a combination of both, as well as by the use of atherogenic Western diets. Early studies in rabbits on an atherogenic diet revealed that a turmeric extract attenuates atherosclerosis development [138]. Similarly, curcumin administration was found to reduce the size of atherosclerotic lesions in a number of mouse models for atherosclerosis [139,140,141,142,143,144].
This anti-atherogenic effect of curcumin could potentially be attributed to its ability to decrease the high plasma cholesterol levels and lipid peroxidation, both of which are fundamental features in the initiation of atherosclerosis, as well as to its capability to change the LDL- to HDL-cholesterol balance towards a more favorable, anti-atherogenic ratio (Figure 3).
“Protective Effects of Curcumin in Cardiovascular Diseases—Impact on Oxidative Stress and Mitochondria” (2022)
In summary, our data show for the first time ROS-mediated HO-1 expression in endothelial cells as a mechanism by which CBD mediates protective autophagy, which at higher CBD concentrations, however, can no longer prevent cell death inducing apoptosis.
“Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy” (2020)
Hyperlipidemia, resulting from the abnormalities of lipid metabolism, is one of the major risk factors for the development of CVD. The elevated levels of plasma lipids such as fatty acids, cholesterol, phospholipids and triglycerides can lead to the development of atherosclerotic plaques [39]. The risk of CVD reduced by 2% with a 1% decrease in serum cholesterol [94] and evidence suggests that drugs with the ability to reduce plasma lipids can reduce the probability of cardiovascular death [39]. Green tea catechins affect lipid metabolism by various mechanisms and prevent the appearance of atherosclerotic plaque in various models of hyperlipidemia [30,88]. In addition, catechins influence micellular solubility, luminal lipid hydrolysis and intestinal lipid absorption [95]. Furthermore, catechins can up-regulate hepatic LDL receptor expression, thereby modulating biosynthesis, excretion and intracellular processing of lipids [20, 95–97].
“Green Tea Catechins and Cardiovascular Health: An Update” (2008)
Oolong tea, partially fermented from Camellia sinensis leaves, exhibits significant antioxidative, anti-inflammatory, and anti-cancer activities as indicated in several in vitro and in vivo studies.
…Oolong tea contains unique polyphenols, such as OFA, which may prevent atherosclerosis by reducing oxidative stress [72]. In apoB-100, cysteine, histidine, lysine, and tyrosine residues are main targets of ROS and RNS, while oxidative modification of apoB-100 may deteriorate the function of apoB-100 as ligand to LDL receptors [72]. The inhibition of peanut oil oxidation and DPPH radical scavenging activity by oolong tea water extracts were mainly associated with polyphenol concentrations, thus emphasizing their preventive role as antioxidants [97]. Noteworthy, the antioxidant activity of oolong tea infusions increased with the elevation of extraction temperature and duration of soaking [97].
…Oxidation of low-density lipoprotein (LDL) in blood by ROS and RNS is often associated with the onset of cardiovascular diseases, such as atherosclerosis. Oolong tea with its unique OFA could reduce lipid peroxidation and oxidative modification of apoB-100 in LDL, therefore preventing atherosclerosis [72].
“Multifunctional health-promoting effects of oolong tea and its products” (2022)
In conclusion, the current study demonstrated that melatonin prevented atherosclerotic progression, at least in part, via inducing mitophagy and attenuating NLRP3 inflammasome activation, which was mediated by the Sirt3/FOXO3a/Parkin signaling pathway. Collectively, our study provides insight into melatonin as a new target for therapeutic intervention for AS.
“Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition” (2018)
Dietary supplementation with polyphenolic antioxidants to animals was shown to be associated with inhibition of LDL oxidation and macrophage foam cell formation, and attenuation of atherosclerosis development. We investigated the effects of pomegranate juice (PJ, which contains potent tannins and anthocyanins) consumption by atherosclerotic patients with carotid artery stenosis (CAS) on the progression of carotid lesions and changes in oxidative stress and blood pressure.
…Blood samples were collected before treatment and during PJ consumption. In the control group that did not consume PJ, common carotid intima-media thickness (IMT) increased by 9% during 1 year, whereas, PJ consumption resulted in a significant IMT reduction, by up to 30%, after 1 year. The patients' serum paraoxonase 1 (PON 1) activity was increased by 83%, whereas serum LDL basal oxidative state and LDL susceptibility to copper ion-induced oxidation were both significantly reduced, by 90% and 59%, respectively, after 12 months of PJ consumption, compared to values obtained before PJ consumption. Furthermore, serum levels of antibodies against oxidized LDL were decreased by 19%, and in parallel serum total antioxidant status (TAS) was increased by 130% after 1 year of PJ consumption. Systolic blood pressure was reduced after 1 year of PJ consumption by 12% [corrected] and was not further reduced along 3 years of PJ consumption. For all studied parameters, the maximal effects were observed after 1 year of PJ consumption. Further consumption of PJ, for up to 3 years, had no additional beneficial effects on IMT and serum PON1 activity, whereas serum lipid peroxidation was further reduced by up to 16% after 3 years of PJ consumption. The results of the present study thus suggest that PJ consumption by patients with CAS decreases carotid IMT and systolic blood pressure and these effects could be related to the potent antioxidant characteristics of PJ polyphenols.
“Pomegranate juice consumption for 3 years by patients with carotid artery stenosis reduces common carotid intima-media thickness, blood pressure and LDL oxidation” (2004)
Fish oil retards the growth of the atherosclerotic plaque by inhibiting both cellular growth factors and the migration of monocytes. The n-3 fatty acids promote the synthesis of the beneficial nitric oxide in the endothelium. Experiments in humans indicate a profound hypolipidemic effect of fish oil, especially lowering of plasma triacylglycerol. Both very-low-density lipoprotein production and apolipoprotein B synthesis are inhibited by fish oil. Finally, fish oil has a mild blood pressure-lowering effect in both normal and mildly hypertensive individuals. These composite effects suggest a prominent therapeutic role for fish oil in the prevention and treatment of coronary artery disease.
“Are fish oils beneficial in the prevention and treatment of coronary artery disease?” (1997)
Fats to limit: Polyunsaturated fatty acids (PUFA’s)
Mostly from vegetable & seed oils, eg peanut, sunflower and rapeseed oil (canola).
Olive, palm oil and coconut fats are the exception.
Crop subsidies ensure over-supply and the food industry is unloading this junk into as much of the food supply as possible. Overconsumption of linoleic acid is problematic as small particle LDL can penetrate the vascular endothelia, oxidise and accelerate pro-inflammatory atherosclerotic pathways.
Do a meta-search and you will find an abundance of flawed studies discussing the fantastic cardiovascular benefits of canola and the healthy vs unhealthy fats argument is a touchpaper for some.
Nothing to see here, move along. Or should that be “moo”ve along, as in BS?
This review is a good start for a more balanced view. Depletion of nitric oxide is associated with vasoconstriction and elevated BP:
Although PUFAs have a protective role against endothelium dysfunction [58], their elevated FFAs adversely affect the endothelium by decreasing NO release and increasing ET-1 levels. Linoleic acid,18:2n-6 (LA) negatively regulates eNOS phosphorylation and affects the intracellular NO levels in ECV304 cells [59].
“Modulation of endothelium function by fatty acids” (2022)
Linoleic acid (LA) is a major constituent of low-density lipoproteins.An essential fatty acid, LA is a polyunsaturated fatty acid, which is oxidised by endogenous enzymes and reactive oxygen species in the circulation.Increased levels of low-density lipoproteins coupled with oxidative stress and lack of antioxidants drive the oxidative processes. This results in synthesis of a range of oxidised derivatives, which play a vital role in regulation of inflammatory processes. The derivatives of LA include, hydroxyoctadecadienoic acids, oxo-octadecadienoic acids, epoxy octadecadecenoic acid and epoxy-keto-octadecenoic acids. In this review, we examine the role of LA derivatives and their actions on regulation of inflammation relevant to metabolic processes associated with atherogenesis and cancer.
…Peroxidation of polyunsaturated fatty acids is intensified in cells subjected to oxidative stress and results in the generation of various bioactive compounds, of which oxidised linoleic acid forms an important component. During the progression of type 2 diabetes mellitus (T2DM), the ensuing hyperglycaemia promotes the generation of reactive oxygen species that contribute to the development of diabetic complications. It has been suggested that reactive oxygen species -induced lipid peroxidation contributes to the development and progression of these pathologies.
…Recent studies have revealed that polytherapy induces an increase in the estimated radius of small, dense LDL lipoprotein subclasses together with a reduction in the level of oxidised LDL particles (Einhorn et al., 2000). These macromolecular rearrangements are associated with changes in the level of specific downstream metabolites produced by linoleic acid peroxidation (i.e. 9-HODE, 13-HODE). Increased HODE levels thus contribute to atherosclerosis progression and the risk of clinical events such as myocardial infarction or stroke.
…In conclusion, a range of derivatives are synthesised from linoleic acid by the action of oxidation or via non-enzymatic pathways. Of these derivatives, predominantly 9- and 13-HODE have been shown to be associated with the regulation of inflammation in metabolic syndrome as well as the processes of cell adhesion, apoptosis, and mitogenesis in cancer (Fig. 2)
“Hydroxyoctadecadienoic acids: Oxidised derivatives of linoleic acid and their role in inflammation associated with metabolic syndrome and cancer” (2015)
The cholesterol lie exposed. Particle size is more critical to CVD outcomes than total cholesterol:
Abstract
Background: The amount of cholesterol per low-density lipoprotein (LDL) particle is variable and related in part to particle size, with smaller particles carrying less cholesterol. This variability causes concentrations of LDL cholesterol (LDL-C) and LDL particles (LDL-P) to be discordant in many individuals.
Methods: LDL-P measured by nuclear magnetic resonance spectroscopy, calculated LDL-C, and carotid intima-media thickness (IMT) were assessed at baseline in the Multi-Ethnic Study of Atherosclerosis, a community-based cohort of 6814 persons free of clinical cardiovascular disease (CVD) at entry and followed for CVD events (n = 319 during 5.5-year follow-up). Discordance, defined as values of LDL-P and LDL-C differing by ≥ 12 percentile units to give equal-sized concordant and discordant subgroups, was related to CVD events and to carotid IMT in models predicting outcomes for a 1 SD difference in LDL-C or LDL-P, adjusted for age, gender, and race.
Results: LDL-C and LDL-P were associated with incident CVD overall: hazard ratios (HR 1.20, 95% CI [CI] 1.08-1.34; and 1.32, 95% CI 1.19-1.47, respectively, but for those with discordant levels, only LDL-P was associated with incident CVD (HR 1.45, 95% CI 1.19-1.78; LDL-C HR 1.07, 95% CI 0.88-1.30). IMT also tracked with LDL-P rather than LDL-C, ie, adjusted mean IMT of 958, 932, and 917 microm in the LDL-P > LDL-C discordant, concordant, and LDL-P < LDL-C discordant subgroups, respectively, with the difference persisting after adjustment for LDL-C (P = .002) but not LDL-P (P = .60).
Conclusions: For individuals with discordant LDL-C and LDL-P levels, the LDL-attributable atherosclerotic risk is better indicated by LDL-P.
“Clinical implications of discordance between low-density lipoprotein cholesterol and particle number” (2011)
Associated with the reduced total HFD intake, IF substantially reduced lesions in the en face aorta and aortic root sinus. It also increased plaque stability by increasing the smooth muscle cell (SMC)/collagen content and fibrotic cap thickness while reducing macrophage accumulation and necrotic core areas. Mechanistically, IF reduced serum total and LDL cholesterol levels by inhibiting cholesterol synthesis in the liver. Meanwhile, HFD-induced hepatic lipid accumulation was attenuated by IF. Interestingly, circulating Ly6Chigh monocytes but not T cells and serum c-c motif chemokine ligand 2 levels were significantly reduced by IF. Functionally, adhesion of monocytes to the aortic endothelium was decreased by IF via inhibiting VCAM-1 and ICAM-1 expression. Taken together, our study indicates that IF reduces atherosclerosis in LDLR-/- mice by reducing monocyte chemoattraction/adhesion and ameliorating hypercholesterolemia and suggests its potential application for atherosclerosis treatment.
“Intermittent Fasting Inhibits High-Fat Diet–Induced Atherosclerosis by Ameliorating Hypercholesterolemia and Reducing Monocyte Chemoattraction” (2021)
Atherosclerosis-associated cardiovascular disease is one of the leading causes of deaths of HIV positive individuals. Key pathologies include stroke and myocardial infarction (MI).
This should alarm us greatly, especially as SARS-CoV-2 and Spike share some of HIV’s cytotoxic envelope proteins due to bioengineering and many of the core pathologies are similar.
This was one of my longest, most referenced Substacks in quite a while but the subject matter is relevant to us all and really cannot have enough attention.
Please bookmark it, share it and keep coming back to it as I add revisions or add your own in the comments.
The next post is likely to be much shorter, partly because I am working on several peer reviewed papers and articles in parallel to this work. Details to follow, its not a quick process.
Key takes from “HIV-1–Associated Atherosclerosis: Unraveling the Missing Link“ by Kearns et al.81 (2018, emphasis mine):
ART: antiretroviral therapy.
There is emerging evidence to indicate that HIV infection and subsequent inflammatory processes in humans accelerate atherogenesis. The study of simian immunodeficiency virus (SIV) infection in primates displays a similar pattern of accelerated atherogenesis, and thus serves as an important model system for HIV pathogenesis.
CVD among HIV+ patients has drastically changed with the advent of ART. Early in the HIV epidemic, the predominant presentations of HIV-associated CVD were dilated cardiomyopathy, pericardial disease, pulmonary hypertension, HIV-associated malignancies, and OIs. Since the onset of ART, HIV+ patients are increasingly at risk for more common cardiovascular complications associated with atherosclerosis, including MI, stroke, and heart failure (2–6).
In patients on ART, HIV infection not only mediates immune cell activation and endothelial dysfunction, but also activates an array of cellular pathways, such as inflammasome formation/caspase-1 activation, autophagy, oxidative stress (OS), and endoplasmic reticulum (ER) stress. These mechanisms have an established contribution in the development of traditional atherosclerosis.
Although fewer HIV+ patients are dying of AIDS-related complications, the prevalence of non-AIDS-related comorbidities, including atherosclerosis-associated CVD, remains increased compared with HIV− controls.
I had no idea before researching for this review:
CVD is the second leading cause of non-AIDS-related mortality in the United States and third in Europe among HIV+ patients (6). Data suggest that HIV+ patients have a higher prevalence of traditional risk factors, including hypertension, diabetes, and dyslipidemia (7).
There is extensive evidence to suggest that even when controlled for these traditional cardiovascular risk factors, HIV+ patients are still at a higher (1.5- to 3-fold) risk of developing CVD (8,9).
HIV+ individuals known as “elite controllers,” that is, those who have undetectable plasma viral loads without ART, have increased coronary atherosclerosis and high immune activation, including elevated plasma soluble CD163 (sCD163) (13). In another cohort, HIV+ elite controllers had a higher median carotid intima-media thickness (CIMT) than that observed in uninfected subjects, even after adjustment for traditional cardiovascular risk factors (14). These studies reinforce the role of inflammation, and not ART or virus, as the key mediator of HIV-associated CVD.
Several HIV proteins are known to contribute to calcium imbalance in endothelial cells and can trigger apoptosis through ER stress, thereby leading to endothelial dysfunction (81). Consistently, HIV Tat-mediated induction of human endothelial cell apoptosis involves ER stress and mitochondrial dysfunction. The HIV envelope protein, gp120, induces type 1 programmed cell death through ER stress using the IRE1α, JNK and AP-1 pathways (33).
DoorlessCarp’s Scientific Literature Reviews is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber. I do not receive any departmental funding, grants for software, online services or equipment etc. It’s your paid subscriptions that make these reviews possible.
Thank you for reading DoorlessCarp’s Scientific Literature Reviews. This post is public so feel free to share it.
This site is strictly an information website reviewing research into potential therapeutic agents. It does not advertise anything, provide medical advice, diagnosis or treatment. This site is not promoting any of these as potential treatments or offers any claims for efficacy. Its content is aimed at researchers, registered medical practitioners, nurses or pharmacists. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website. Always consult a qualified health provider before introducing or stopping any medications as any possible drug interactions or effects will need to be considered.
Any extracts used in the previous article are for non-commercial research and educational purposes only and may be subject to copyright from their respective owners.
L ópez MB, Fern ández FL, Oliva B, et al. Short-Term Progression of Multiterritorial Subclinical Atherosclerosis.Journal of the American College of Cardiology. 2020;75(14):1617-1627. doi:10.1016/j.jacc.2020.02.026
Zaman AG, Helft G, Worthley SG, Badimon JJ. The role of plaque rupture and thrombosis in coronary artery disease.Atherosclerosis. 2000;149(2):251-266. doi:10.1016/s0021-9150(99)00479-7
Ren S, Fan X, Peng L, et al. Expression of NF-κB, CD68 and CD105 in carotid atherosclerotic plaque.J Thorac Dis. 2013;5(6):771-776. doi:10.3978/j.issn.2072-1439.2013.12.36
Thakur A, Sharma V, Averbek S, et al. Immune landscape and redox imbalance during neurological disorders in COVID-19.Cell Death Dis. 2023;14(9):1-12. doi:10.1038/s41419-023-06102-6
Shah P, Bajaj S, Virk H, Bikkina M, Shamoon F. Rapid Progression of Coronary Atherosclerosis: A Review.Thrombosis. 2015;2015:634983. doi:10.1155/2015/634983
Perico L, Benigni A, Remuzzi G. SARS-CoV-2 and the spike protein in endotheliopathy.Trends in Microbiology. 2024;32(1):53-67. doi:10.1016/j.tim.2023.06.004
Dilliard SA, Cheng Q, Siegwart DJ. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles.Proc Natl Acad Sci U S A. 2021;118(52):e2109256118. doi:10.1073/pnas.2109256118
Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery.J Control Release. 2006;114(1):100-109. doi:10.1016/j.jconrel.2006.04.014
Pradhan P, Pandey A, Mishra A, et al. Uncanny Similarity of Unique Inserts in the 2019-nCoV Spike Protein to HIV-1 Gp120 and Gag.; 2020. doi:10.1101/2020.01.30.927871
Curlin J, Schmitt K, Remling-Mulder L, et al. Evolution of SIVsm in humanized mice towards HIV-2.J Med Primatol. 2020;49(5):280-283. doi:10.1111/jmp.12486
Milosevits G, Szebeni J, Krol S. Exosomes: potential model for complement-stealth delivery systems.European Journal of Nanomedicine. 2015;7(3):207-218. doi:10.1515/ejnm-2015-0005
Bansal S, Perincheri S, Fleming T, et al. Cutting Edge: Circulating Exosomes with COVID Spike Protein Are Induced by BNT162b2 (Pfizer-BioNTech) Vaccination prior to Development of Antibodies: A Novel Mechanism for Immune Activation by mRNA Vaccines.J Immunol. 2021;207(10):2405-2410. doi:10.4049/jimmunol.2100637
Hanna N, Mejia CMD, Heffes-Doon A, et al. Biodistribution of mRNA COVID-19 vaccines in human breast milk.eBioMedicine. 2023;96. doi:10.1016/j.ebiom.2023.104800
Vilaplana-Carnerero C, Giner-Soriano M, Dominguez À, et al. Atherosclerosis, Cardiovascular Disease, and COVID-19: A Narrative Review.Biomedicines. 2023;11(4):1206. doi:10.3390/biomedicines11041206
Vidarsson G, Dekkers G, Rispens T. IgG Subclasses and Allotypes: From Structure to Effector Functions.Front Immunol. 2014;5:520. doi:10.3389/fimmu.2014.00520
Liu Y, Zhang HG. Vigilance on New-Onset Atherosclerosis Following SARS-CoV-2 Infection.Front Med (Lausanne). 2021;7:629413. doi:10.3389/fmed.2020.629413
Tougaard NH, Theilade S, Winther SA, et al. Carotid‐Femoral Pulse Wave Velocity as a Risk Marker for Development of Complications in Type 1 Diabetes Mellitus.J Am Heart Assoc. 2020;9(19):e017165. doi:10.1161/JAHA.120.017165
Fan J, Li X, Zhong L, et al. MCP-1, ICAM-1 and VCAM-1 are present in early aneurysmal dilatation in experimental rats.Folia Histochem Cytobiol. 2010;48(3):455-461. doi:10.2478/v10042-010-0042-y
Lee AR, Woo JS, Lee SY, et al. SARS-CoV-2 spike protein promotes inflammatory cytokine activation and aggravates rheumatoid arthritis.Cell Communication and Signaling. 2023;21(1):44. doi:10.1186/s12964-023-01044-0
Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix Metalloproteinase-9: Many Shades of Function in Cardiovascular Disease. Physiology (Bethesda). 2013;28(6):391-403. doi:10.1152/physiol.00029.2013
Roy A, Sarkar A, Nayak D, Das S. Ultradiluted SARS-CoV-2 Spike Protein mitigates hyperinflammation in lung via ferritin and MMP-9 regulation in BALB/c mice.Virus Res. 2023;329:199091. doi:10.1016/j.virusres.2023.199091
Ren M, Wang T, Wei X, et al. LncRNA H19 regulates smooth muscle cell functions and participates in the development of aortic dissection through sponging miR-193b-3p.Biosci Rep. 2021;41(1):BSR20202298. doi:10.1042/BSR20202298
Moazzam‐Jazi M, Lanjanian H, Maleknia S, Hedayati M, Daneshpour MS. Interplay between SARS‐CoV‐2 and human long non‐coding RNAs.J Cell Mol Med. 2021;25(12):5823-5827. doi:10.1111/jcmm.16596
Heo JY, Seo YB, Kim EJ, et al. COVID-19 vaccine type-dependent differences in immunogenicity and inflammatory response: BNT162b2 and ChAdOx1 nCoV-19.Frontiers in Immunology. 2022;13. Accessed January 27, 2024. https://www.frontiersin.org/articles/10.3389/fimmu.2022.975363
Liwsrisakun C, Pata S, Laopajon W, et al. Neutralizing antibody and T cell responses against SARS-CoV-2 variants of concern following ChAdOx-1 or BNT162b2 boosting in the elderly previously immunized with CoronaVac vaccine. Immunity & Ageing. 2022;19(1):24. doi:10.1186/s12979-022-00279-8
Natarelli L, Parca L, Mazza T, Weber C, Virgili F, Fratantonio D. MicroRNAs and Long Non-Coding RNAs as Potential Candidates to Target Specific Motifs of SARS-CoV-2.Noncoding RNA. 2021;7(1):14. doi:10.3390/ncrna7010014
Yao XP, Ye J, Feng T, et al. Adaptor protein MyD88 confers the susceptibility to stress via amplifying immune danger signals.Brain Behav Immun. 2023;108:204-220. doi:10.1016/j.bbi.2022.12.007
Sahanic S, Hilbe R, Dünser C, et al. SARS-CoV-2 activates the TLR4/MyD88 pathway in human macrophages: A possible correlation with strong pro-inflammatory responses in severe COVID-19.Heliyon. 2023;9(11):e21893. doi:10.1016/j.heliyon.2023.e21893
Zheng M, Karki R, Williams EP, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines.Nat Immunol. 2021;22(7):829-838. doi:10.1038/s41590-021-00937-x
Sheahan T, Morrison TE, Funkhouser W, et al. MyD88 Is Required for Protection from Lethal Infection with a Mouse-Adapted SARS-CoV.PLOS Pathogens. 2008;4(12):e1000240. doi:10.1371/journal.ppat.1000240
Chu T, Xu X, Ruan Z, Wu L, Zhou M, Zhu G. miR-146a contributes to atherosclerotic plaque stability by regulating the expression of TRAF6 and IRAK-1.Mol Biol Rep. 2022;49(6):4205-4216. doi:10.1007/s11033-022-07253-z
Lundberg AM, Ketelhuth DFJ, Johansson ME, et al. Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis.Cardiovascular Research. 2013;99(2):364-373. doi:10.1093/cvr/cvt033
Liang S, Bao C, Yang Z, et al. SARS-CoV-2 spike protein induces IL-18-mediated cardiopulmonary inflammation via reduced mitophagy.Sig Transduct Target Ther. 2023;8(1):1-15. doi:10.1038/s41392-023-01368-w
Messerli FH, Panjrath GS. The J-curve between blood pressure and coronary artery disease or essential hypertension: exactly how essential?J Am Coll Cardiol. 2009;54(20):1827-1834. doi:10.1016/j.jacc.2009.05.073
Andersson OK, Almgren T, Persson B, Samuelsson O, Hedner T, Wilhelmsen L. Survival in treated hypertension: follow up study after two decades.BMJ. 1998;317(7152):167-171. Accessed February 1, 2024. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC28606/
Dykun I, Lehmann N, Kälsch H, et al. Statin Medication Enhances Progression of Coronary Artery Calcification: The Heinz Nixdorf Recall Study.J Am Coll Cardiol. 2016;68(19):2123-2125. doi:10.1016/j.jacc.2016.08.040
Henein M, Granåsen G, Wiklund U, et al. High dose and long-term statin therapy accelerate coronary artery calcification.Int J Cardiol. 2015;184:581-586. doi:10.1016/j.ijcard.2015.02.072
Svensson P, de Faire U, Sleight P, Yusuf S, Östergren J. Comparative Effects of Ramipril on Ambulatory and Office Blood Pressures.Hypertension. 2001;38(6):e28-e32. doi:10.1161/hy1101.099502
Kannel WB. Risk stratification in hypertension: new insights from the Framingham Study.Am J Hypertens. 2000;13(1 Pt 2):3S-10S. doi:10.1016/s0895-7061(99)00252-6
Krysiak R, Okopień B. Pleiotropic effects of angiotensin-converting enzyme inhibitors in normotensive patients with coronary artery disease.Pharmacol Rep. 2008;60(4):514-523.
Ahn SA, Jong P, Pouleur H, Rousseau MF. Abstract 17212: Effect of Enalapril on Long-Term Survival among Patients with Left Ventricular Systolic Dysfunction: a 25-Year Near-Full Life-Cycle Follow-Up of SOLVD in Belgium.Circulation. 2014;130(suppl_2):A17212-A17212. doi:10.1161/circ.130.suppl_2.17212
HOPE/HOPE-TOO Study Investigators. Long-Term Effects of Ramipril on Cardiovascular Events and on Diabetes.Circulation. 2005;112(9):1339-1346. doi:10.1161/CIRCULATIONAHA.105.548461
Pullarkat V, Cruz-Chacon A, Gangatharan S, et al. P1070: NAVITOCLAX MONOTHERAPY IN PATIENTS WITH MYELOFIBROSIS PREVIOUSLY TREATED WITH JAK-2 INHIBITORS: SAFETY AND TOLERABILITY.Hemasphere. 2022;6(Suppl):960-961. doi:10.1097/01.HS9.0000847148.78233.c8
Kearns A, Gordon J, Burdo TH, Qin X. HIV-1–Associated Atherosclerosis: Unraveling the Missing Link.J Am Coll Cardiol. 2017;69(25):3084-3098. doi:10.1016/j.jacc.2017.05.012
One in three health workers suffering ‘burnout’ amid NHS staffing crisis
...high blood pressure, chest pains and headaches are among the physical signs of stress reported by nurses, porters, 999 call handlers and other NHS staff who completed the survey.







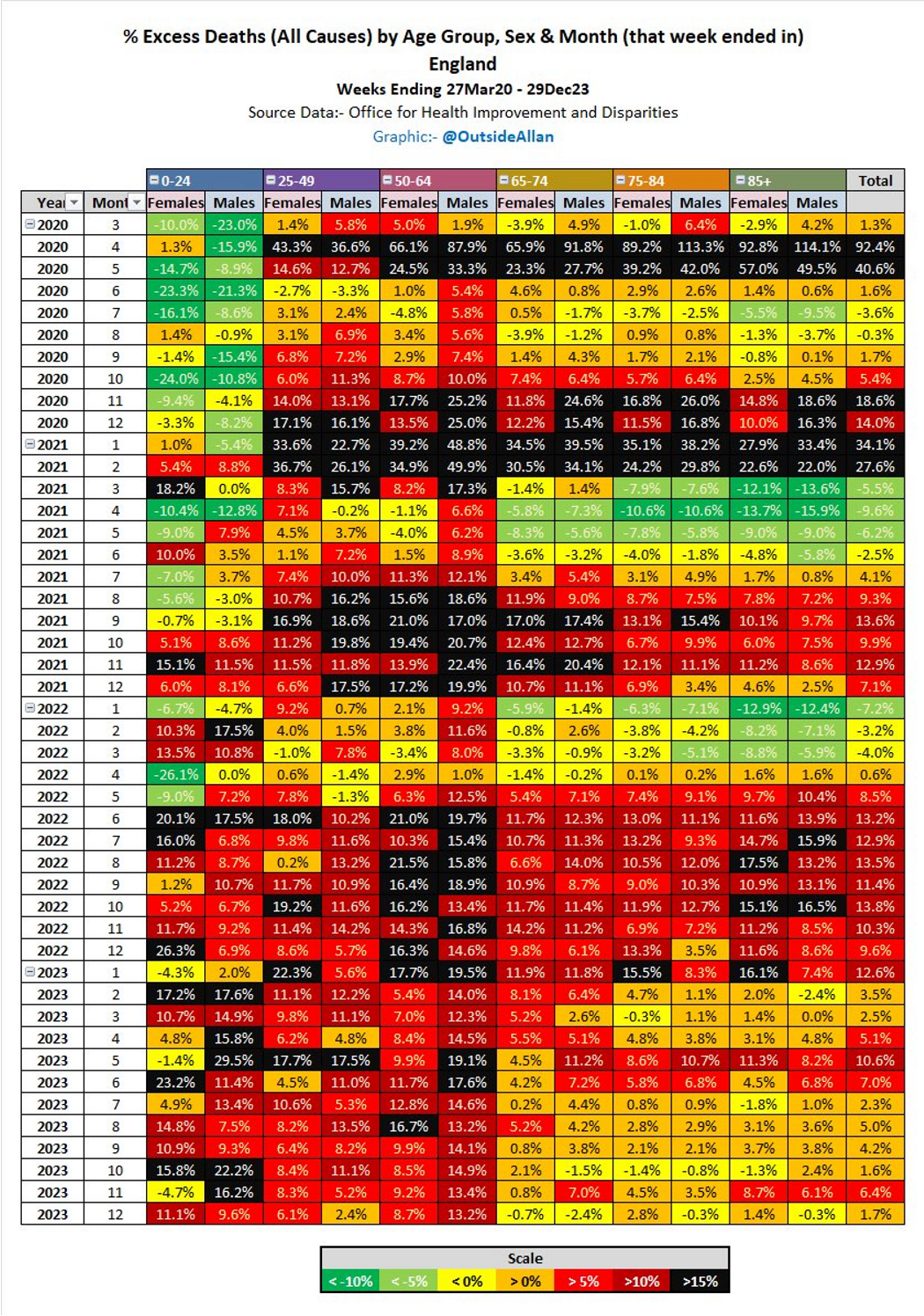
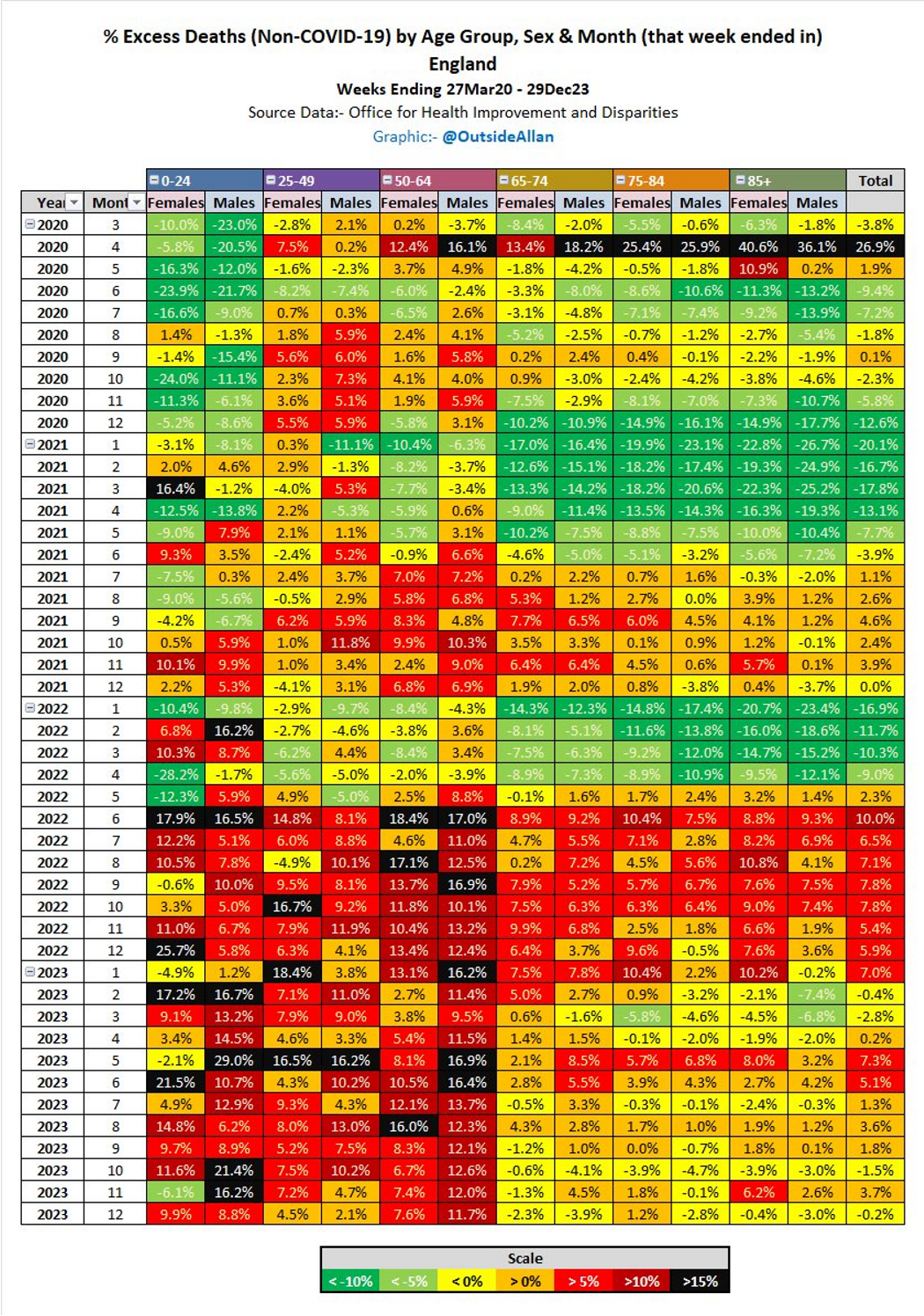
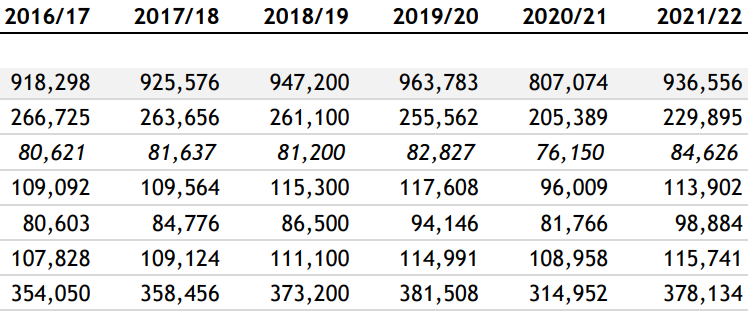
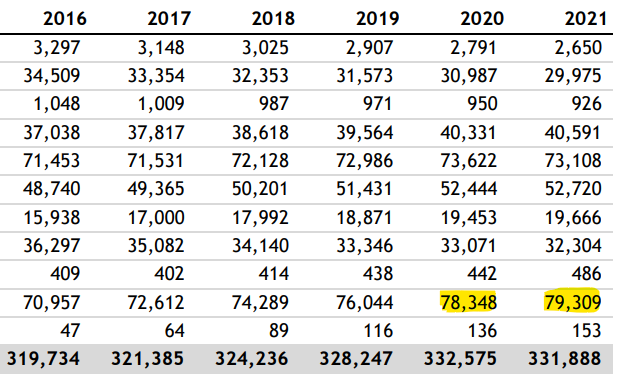
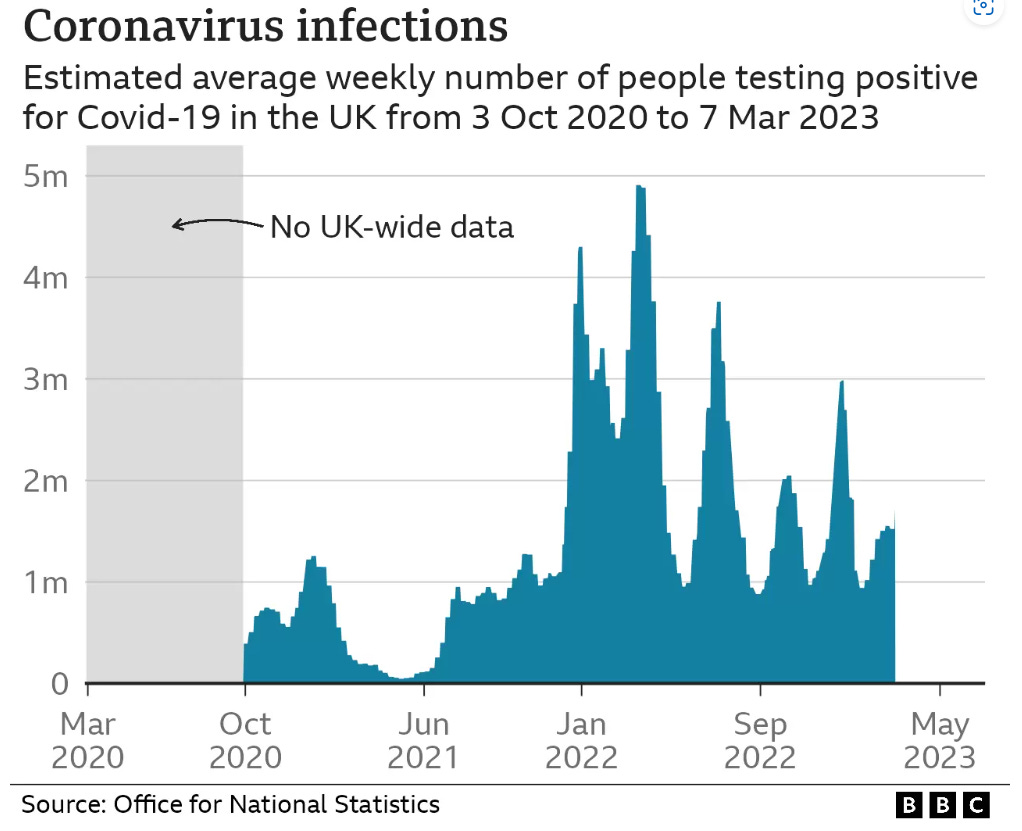
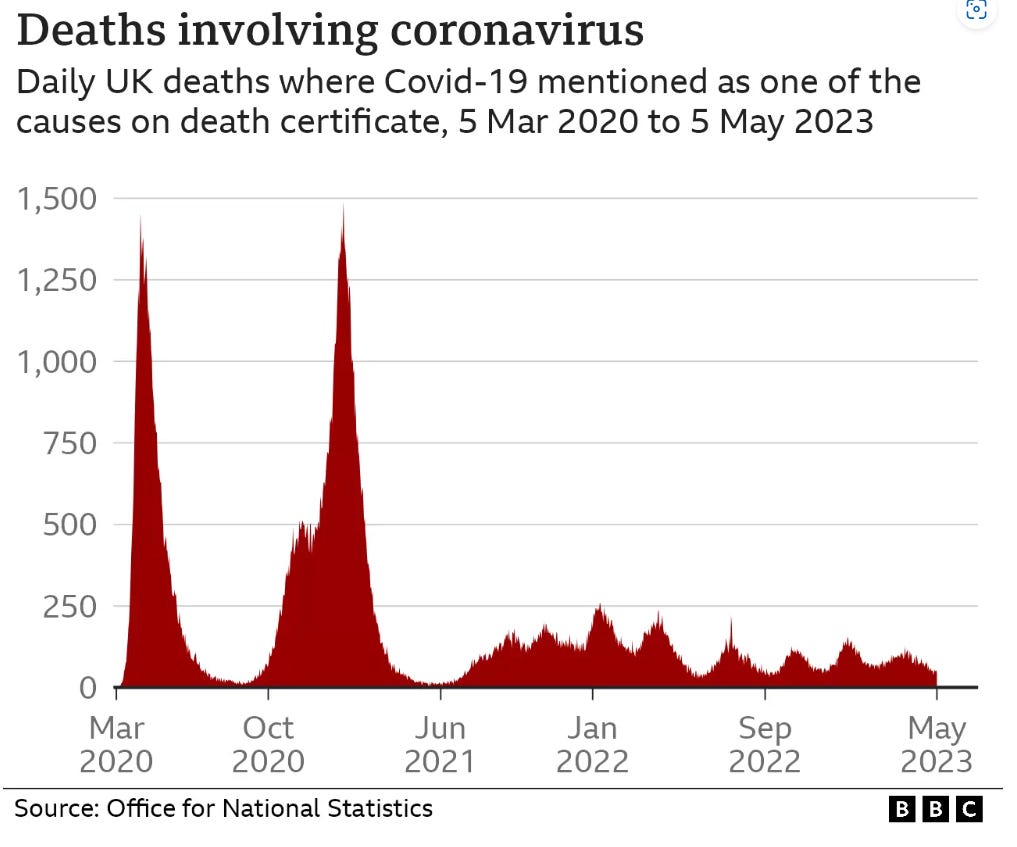


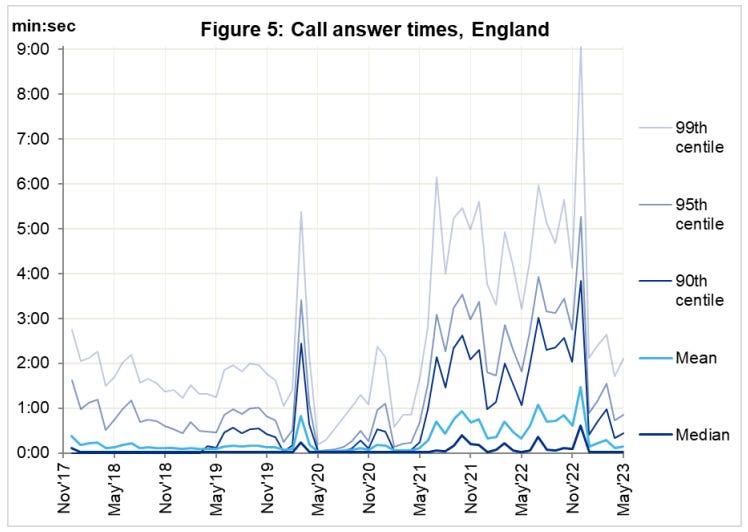











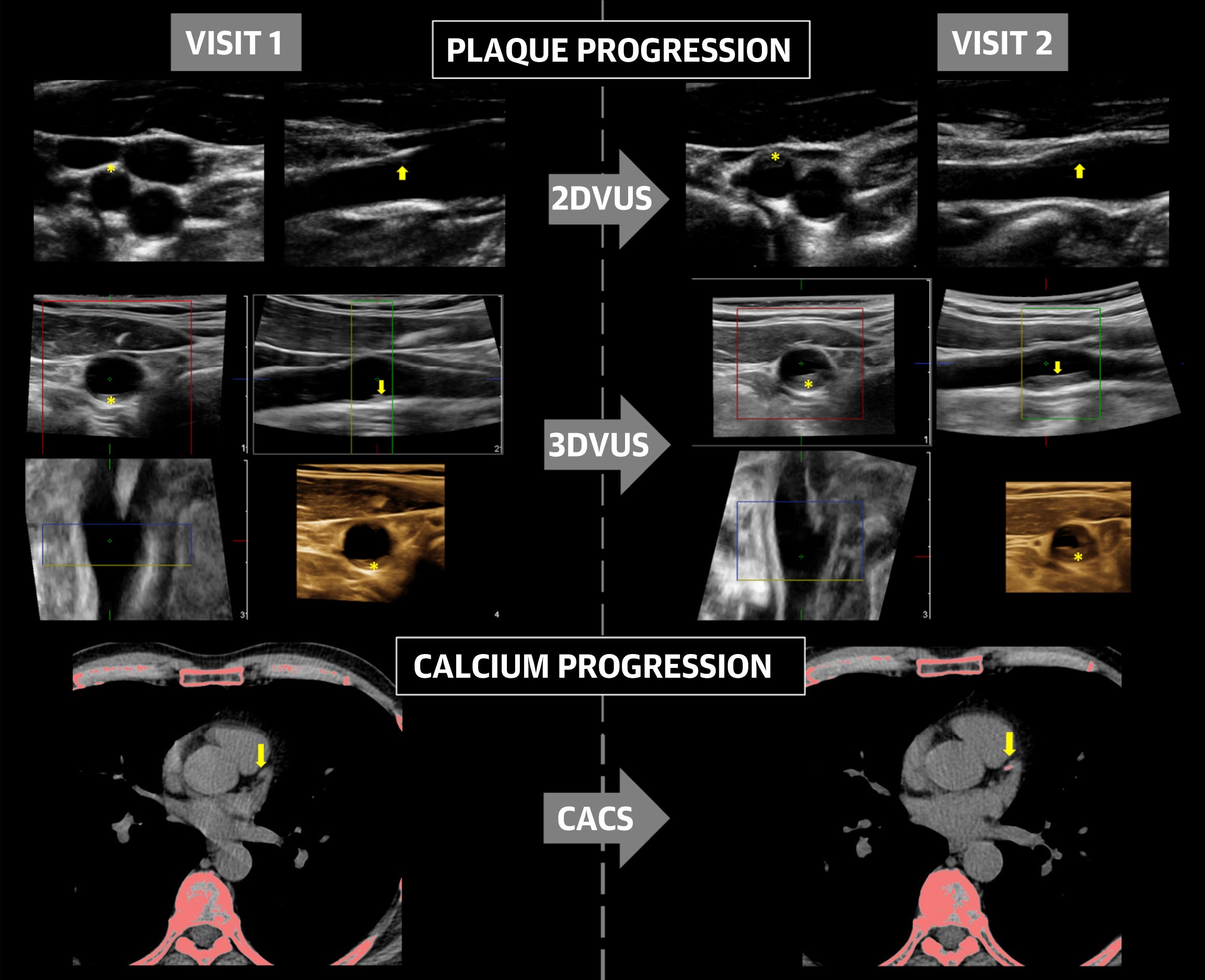





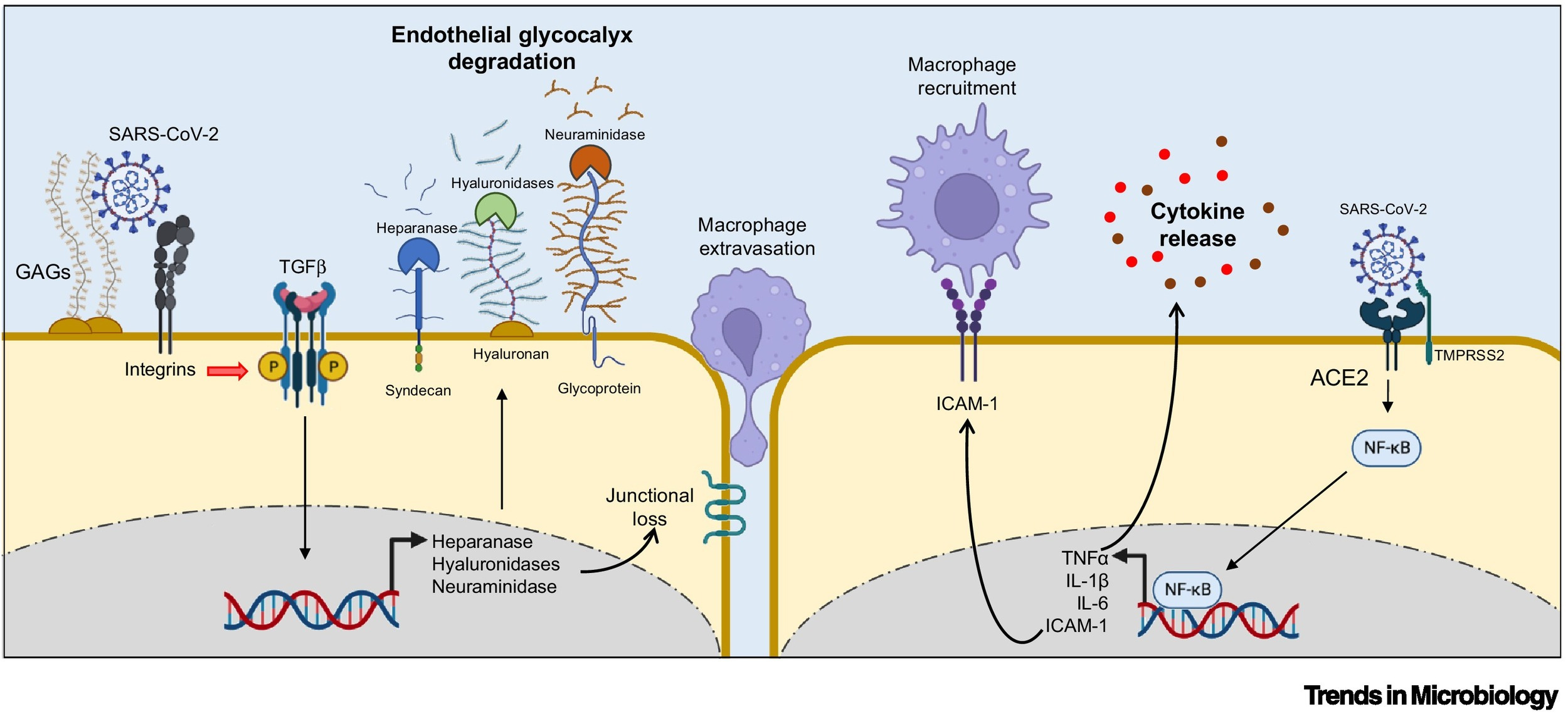



.")

.")

 Representative NanoSight image for exosomes from vaccinated individuals with mean and median sizes (black thin line in the graph indicates the three measurements of the same sample, and red line is the average of all three lines). (B) Transmission electron microscopy images of SARS-CoV-2 spike Ag on exosomes from control exosomes from control and vaccinated individuals. Arrows indicate SARS-CoV-2 spike-positive exosomes. Right side, third image is the zoomed image of positive exosome from vaccinated sample (original magnification x 50,000). We have used anti-coronavirus FIPV3-70 Ab as negative control for both the samples.")
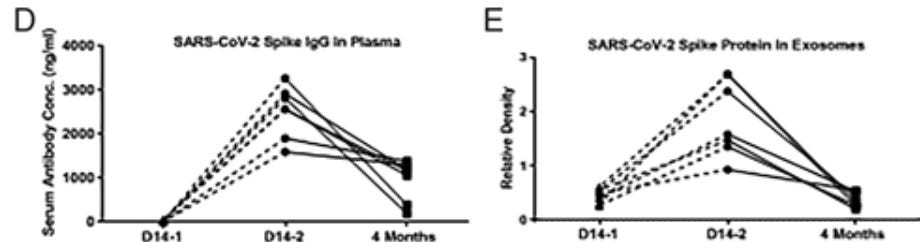


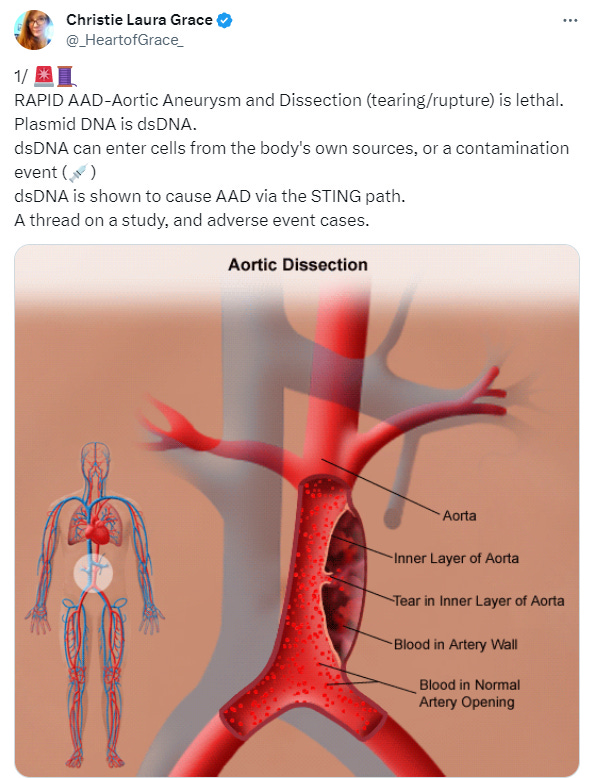





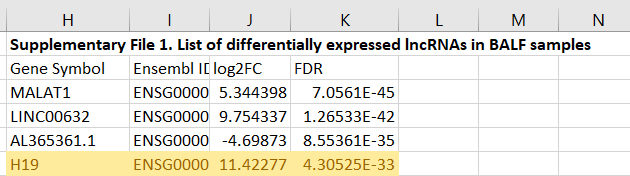







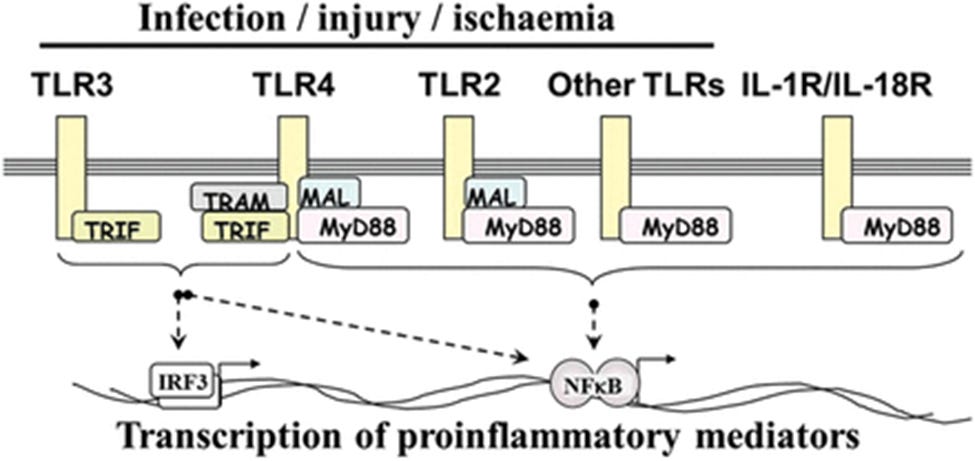

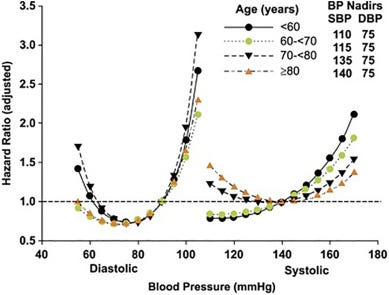
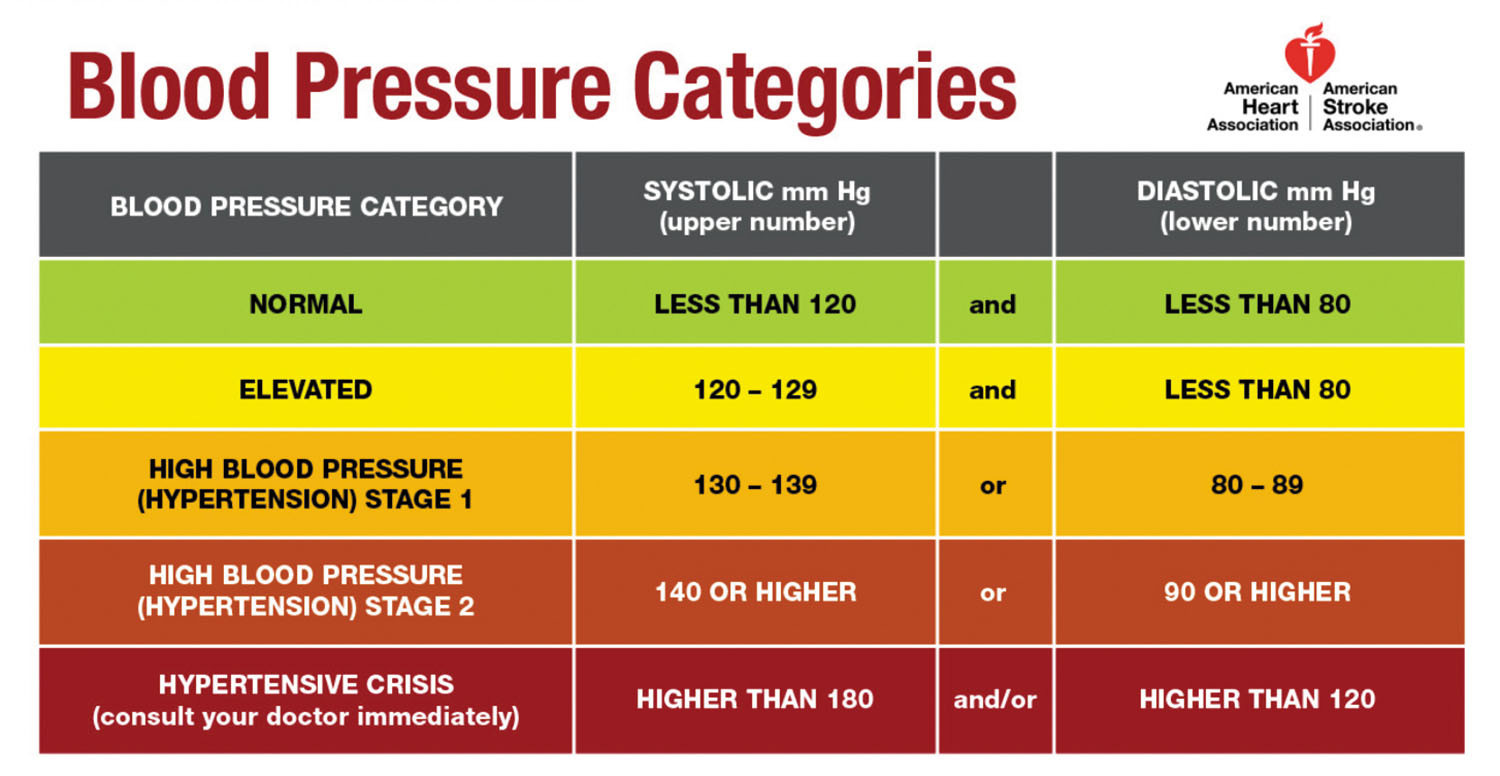



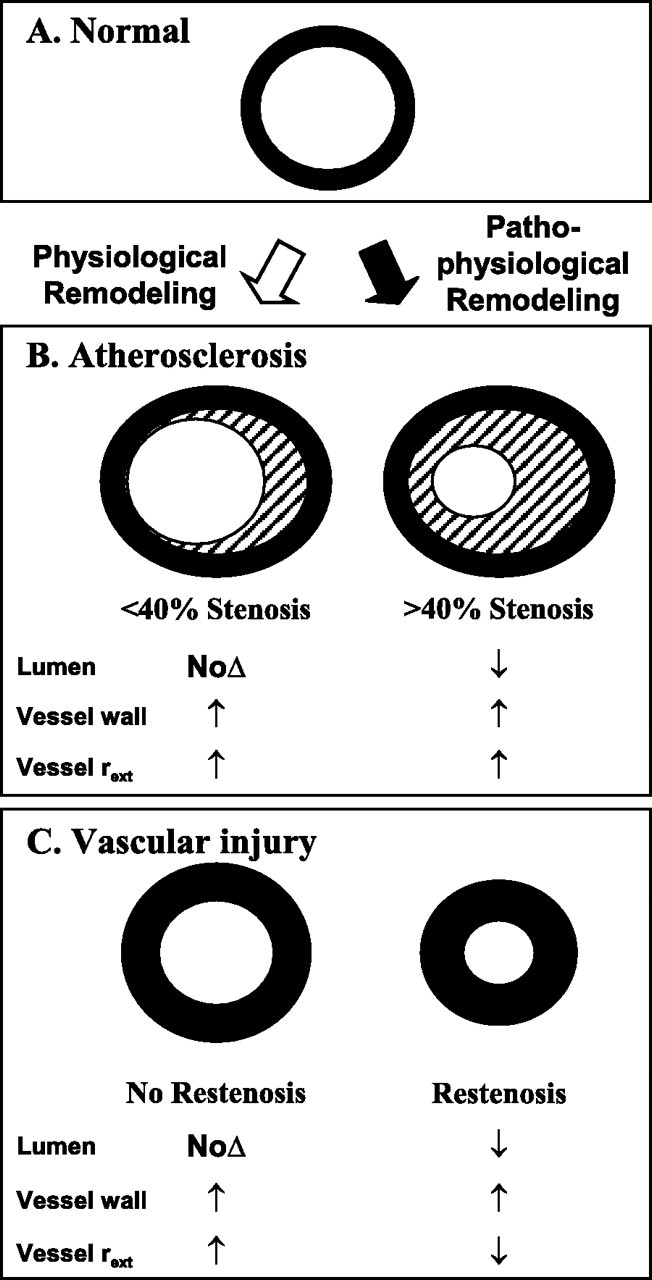
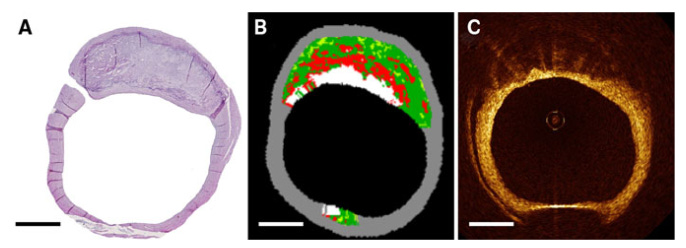
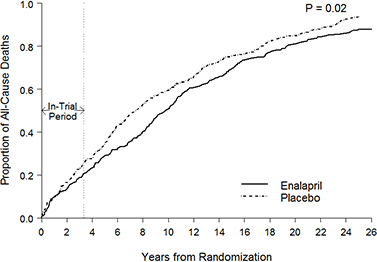
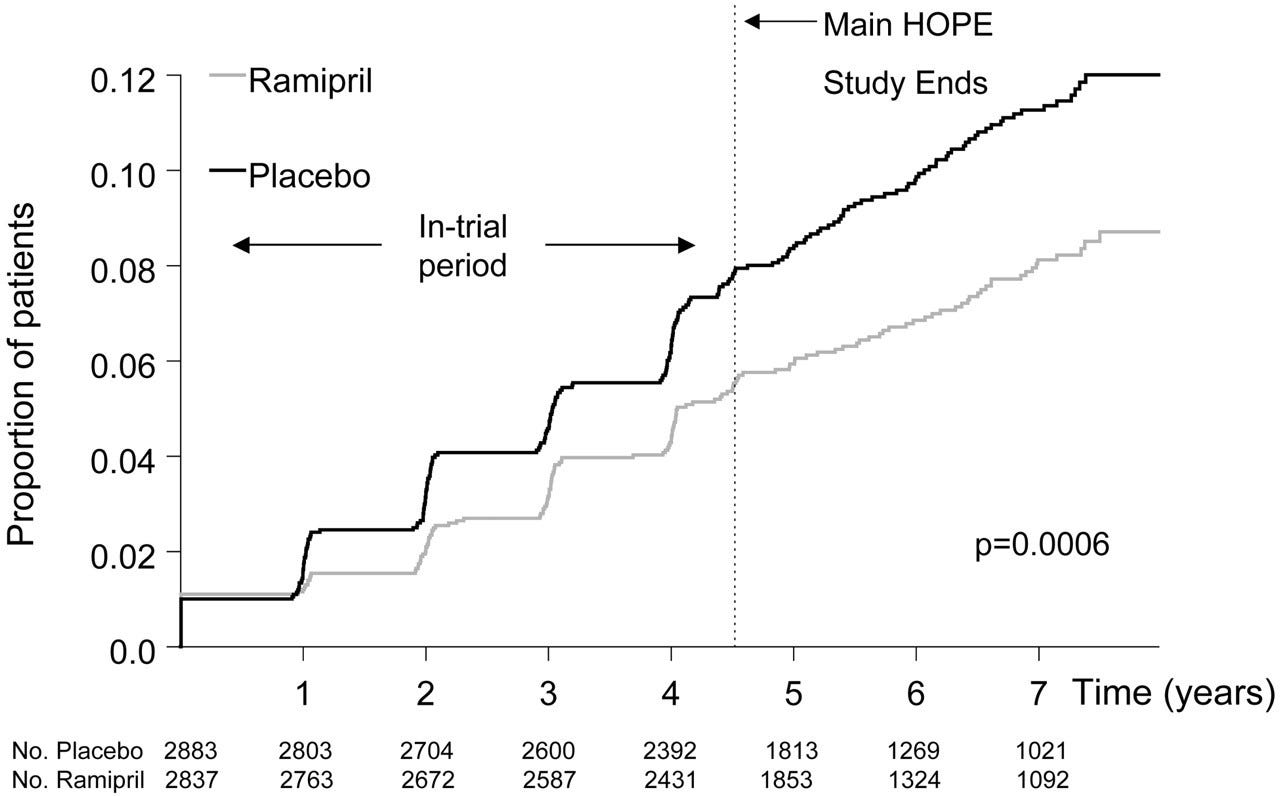



{kind=link}
 - S01E14 Go Away Ghost Ship | Video gifs by quotes | 5994f1e8 | 紗")

Gaslighting themselves?
One in three health workers suffering ‘burnout’ amid NHS staffing crisis
...high blood pressure, chest pains and headaches are among the physical signs of stress reported by nurses, porters, 999 call handlers and other NHS staff who completed the survey.
https://www.msn.com/en-gb/health/other/one-in-three-health-workers-suffering-burnout-amid-nhs-staffing-crisis/ar-BB1lgvuJ
Background:
Vaccination as a condition of deployment (VCOD) for healthcare workers: frequently asked questions
https://www.england.nhs.uk/coronavirus/documents/vaccination-as-a-condition-of-deployment-vcod-for-healthcare-workers-frequently-asked-questions/
One of your best yet. An absolute tour de force.